Apoptosome
The apoptosome is a large quaternary protein structure formed in the process of apoptosis. Its formation is triggered by the release of cytochrome c from the mitochondria in response to an internal (intrinsic) or external (extrinsic) cell death stimulus. Stimuli can vary from DNA damage and viral infection to developmental cues such as those leading to the degradation of a tadpole's tail.
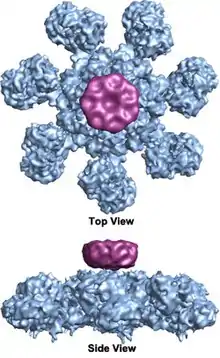
In mammalian cells, once cytochrome c is released, it binds to the cytosolic protein Apaf-1 to facilitate the formation of an apoptosome. An early biochemical study suggests a two-to-one ratio of cytochrome c to apaf-1 for apoptosome formation. However, recent structural studies suggest the cytochrome c to apaf-1 ratio is one-to-one. It has also been shown that the nucleotide dATP as third component binds to apaf-1, however its exact role is still debated. The mammalian apoptosome had never been crystallized, but a human APAF-1/cytochrome-c apoptosome has been imaged at lower (2 nm) resolution by cryogenic transmission electron microscopy in 2002,[2] revealing a heptameric wheel-like particle with 7-fold symmetry. Recently, a medium resolution (9.5 Ångström) structure of human apoptosome was also solved by cryo-electron microscopy, which allows unambiguous inference for positions of all the APAF-1 domains (CARD, NBARC and WD40) and cytochrome c. There is also now a crystal structure of the monomeric, inactive Apaf-1 subunit (PDB 3SFZ).[1][3]
Once formed, the apoptosome can then recruit and activate the inactive pro-caspase-9. Once activated, this initiator caspase can then activate effector caspases and trigger a cascade of events leading to apoptosis.
History
The term Apoptosome was first introduced in Yoshihide Tsujimoto's 1998 paper "Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria?".[4] However, the Apoptosome was known before this time as a ternary complex. This complex involved caspase-9 and Bcl-XL which each bound a specific Apaf-1 domain. The formation of this complex was then believed to play a regulatory role in mammalian cell death.[5] In December of the same year, a further article was released in The Journal of Biological Chemistry stating that Apaf-1 is the regulator of apoptosis, through activation of procaspase-9.[6]
The criteria for an apoptosome were laid out in 1999. Firstly, it must be a large complex (greater than 1.3 million Daltons). Secondly its formation requires the hydrolysis of a high energy bond of ATP or dATP. And lastly it must activate procaspase-9 in its functional form. The formation of this complex is the point of no return, and apoptosis will occur. The stable APAF-1 and cytochrome mutimeric complex fit this description, and is now called the apoptosome.[7]
The apoptosome was thought to be a mutimeric complex for two reasons. Firstly, to bring multiple procaspase-9 molecules close together for cleavage. And secondly, to raise the threshold for apoptosis, therefore nonspecific leakage of cytochrome c would not result in apoptosis.[7]
Once the apoptosome was established as the procaspase-9 activator, mutations within this pathway became an important research area. Some examples include human leukemia cells, ovarian cancer and viral infections.[8][9][10] Current research areas for this pathway will be discussed in further detail. There are hidden routes for cell death as well, which are independent of APAF-1 and therefore the apoptosome. These routes are also independent of caspase-3 and 9. These hidden pathways for apoptosis are slower, but may prove useful with further research.[11]
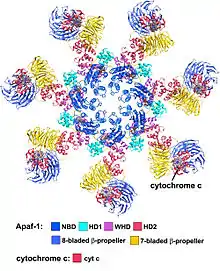
(Yuan et al. 2010, Structure of an apoptosome-procaspase-9 CARD complex[1]
Structure
The apoptosome is a multimolecular holoenzyme complex assembled around the adaptor protein Apaf1 (apoptotic protease activating factor 1) upon mitochondria-mediated apoptosis which must be stimulated by some type of stress signal T formation of the apoptosome requires the presence of ATP/dATP and cytochrome c in the cytosol.[12] A stress stimulus can trigger the release of cytochrome c into the cytoplasm which will then bind to the C-terminus of Apaf-1 within a region containing multiple WD-40 repeats.[2] The oligomerization of Apaf-1 appears to be accompanied by synchronized recruitment of procaspase-9 to the CARD motif at the Apaf-1 N-terminus.[2] The apoptosome triggers the activation of caspases in the intrinsic pathway of apoptosis.[12]
The wheel-shaped heptameric complex with sevenfold symmetry structure of the apoptosome was first revealed at 27 Å resolution by electron cryomicroscopy techniques and has a calculated mass of about 1 MDa (Acehan et al. 2002).[2] This wheel-like particle has seven spokes and a central hub. The distal region of the spoke has a pronounced Y shape.[12] The hub domain is connected to the Y domain by a bent arm. Each Y domain comprises two lobes (a large one and a small one) between which cytochrome c binding sites.[12] Because the resolution of the apoptosome structure was relatively low, two controversial models for apoptosome assembly were proposed. One model suggests NOD domains form the central hub and the CARD domains form a freer ring at the top of the NOD region.[2] Another model proposes that Apaf-1 is organized in an extended fashion such that both the N-terminal CARD and the nucleotide binding region form the central hub of the apoptosome, whereas the 13 WD-40 repeats constitute the two lobes.[12] The large lobe is formed by seven repeats and the small lobe is formed by six repeats.[12] Each caspase- 9 molecule binds a CARD domain at the central hub, forming a dome shaped structure.[12] This controversy has been resolved by a recent high resolution structure of the human apoptosome-procaspase-9 CARD complex.[1] This structure clearly demonstrated that only the NOD regions form the central hub of the apoptosome (see pictures), while CARD is flexibly linked to the platform of apoptosome and becomes disordered in the ground state apoptosome.[1] Once apoptosome binds to procaspase-9, the Apaf-1 CARDs and procaspase-9 CARDs form a flexible disk-like structure sitting above the platform.[1] The number of WD-40 repeats has also been proved to be 15 instead of 13,[1] and it is composed of a 7-bladed beta-propeller and an 8-bladed beta-propeller.[1]
Evidence from Wang and colleagues indicates that the stoichiometric ratio of procaspase-9 to Apaf-1 within the complex is approximately 1:1 .[7] This was further proved by quantitative mass spectrometry analysis.[13] The stoichiometry of cytochrome c to Apaf-1 within the complex is proved to be 1:1.[1] There are some debates about whether stable incorporation of cytochrome c into the apoptosome is required following oligomerization, but recent structural data favor the idea that cytochrome c stabilizes the oligomeric human apoptosome.[1] However, cytochrome c may be not required for the assembly of apoptosome in non-mammalian species, such as worms and fruit flies.[14] In addition, several other molecules, most notably caspase-3, have been reported to co-purify with the apoptosome[7] and caspase-3 has been proved to be able to bind the apoptosome-procaspase-9 complex.[13]
Apaf-1 forms the backbone of the apoptosome. It has three distinct regions: the N-terminal caspase-recruitment domain (CARD, residues 1–90), a central nucleotide-binding and oligomerization region (NB-ARC/NOD, 128–586) and a C-terminal WD40 region (613–1248) making up a protein about 140 KDa.[2]
- The CARD domain of Apaf-1 interacts with procaspase-9 and involved with recruitment within the apoptosome.[2]
- The NB-ARC/NOD region exhibits significant sequence similarity to the C. elegans Ced-4 protein.[2]
- The C-terminal WD40 region of Apaf1 contains 15 WD-40 repeats structured into two b-propeller-shaped domains.[1] WD-40 repeats are sequences around 40 amino acids long which end in Trp-Asp and are typically involved in protein–protein interaction.[2]
A short linker and nucleotide binding a/b domains (NBD) that contain conserved Walker boxes A (p-loop 155-161) and B (239-243) follow the N-terminal CARD domain.[2] The Walker boxes A/B are critical for dATP/ATP and Mg2+ binding.[1][2] Following the NBD is a small helical domain (HD1), a second linker and a conserved winged helix domain (WHD).[2] The NOD region comprises NBD, HD1 and WHD, creating an ATPase domain that is part of the AAA+ family of ATPases.[1][2] There is a super helical domain (HD2) present in the junction between the NOD and the WD-40 repeats.[1] The WD40 repeats are in groups of eight and seven with linkers connecting them.[1]
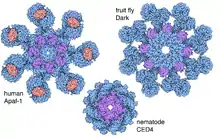
Apoptosomes in other organisms
The above descriptions are for the human apoptosome. Apoptosome complex structures from other organisms have many similarities, but are of quite different sizes and numbers of subunits, as shown in the figure. The fruit-fly system, called Dark, has a ring of 8 subunits (PDB 4V4L).[14] The nematode apoptosome, called CED-4, is octameric but much smaller (PDB 3LQQ), and it does not include the regions that would bind cytochrome C.[15]
Mechanism of Action
Initiation
The initiation of apoptosome action corresponds with the first steps in the programmed cell death (PCD) pathway. In animals, apoptosis can be catalyzed in one of two ways; the extrinsic pathway involves binding of extracellular ligands to transmembrane receptors, while the intrinsic pathway take place in the mitochondria.[16] This intrinsic pathway involves the release of cytochrome C from the mitochondria and subsequent binding to the cytosolic protein Apaf-1.[16][17] Cytochrome c release is thus necessary for the initiation of apoptosome action; this release is regulated in a number of ways, most importantly by detection of calcium ion levels.[16]
Cytochrome c Release
Cytochrome c release is proposed to take place in one of two ways. Firstly, the permeability transition pore (PTP) when the mitochondria receives a death inducing signal, and releases intermembrane space proteins (12). The PTP is composed of the voltage-dependent anion channel (VDAC), the inner membrane protein adenine nucleotide translocator (AdNT) and the matrix protein cyclophilin D (CyD) (12). This pore causes the mitochondria to swell and the outer mitochondrial membrane to rupture (Diamond & McCabe, 2007). With this change in permeability, proteins such as cytochrome c are released into the cytosol (12). This change likely causes the mitochondrial permeability transition (MPT), where the mitochondrial transmembrane potential collapses, and ATP production ceases (12). The inhibition of this method by the pharmaceutical agent cyclosporine A (CsA), lead to the discovery of the second pathway (13). The second method of cytochrome c release is independent of the PTP and involves only the VDAC. Members of the Bcl-2 family of pro-apoptotic proteins can induce the opening of the VDAC (12). This will cause the same release of intermembrane space proteins, including cytochrome c, and the subsequent MPT to occur (12).
a. Absence of Cytochrome c
In the absence of cytochrome c, Apaf-1 exists in its monomeric form; it is thought that the WD-40 domain remain folded back onto the protein, keeping Apaf-1 in an auto inhibited state.[16] In addition, several areas are so tightly bound that the protein is unable to bind to anything else.[16] It has been determined through mass spectrometry that in the autoinhibited, or "locked" state, ADP is bound to the ATPase domain of Apaf-1.[16] In this state, this protein is singular, and incapable of activating any caspases.
b. Presence of Cytochrome c
Cytochrome c binds to the WD-40 domain of Apaf-1.[16] This allows for the "lock" to be released, meaning this domain is no longer autoinhibited.[1][16] However, the CARD and NB-ARC domains remain in autoinhibited state.[16] The CARD domain will only be released from this lock when Apaf-1 is bound to (d) ATP/ATP; when ATP binds, the CARD domain will then be allowed to bind to Caspase-9.[1][16] When ADP is in the ATPase domain, oligomerization is inhibited. Thus, the binding of ATP also allows for the oligomerization of Apaf-1 into the heptagonal structure necessary for downstream caspase activation.[1][7][16] Mutations in the ATPase domain render the protein inactive; however, the method of controlling this ADP-ATP exchange is unclear.[1][7][16] Oligomerization can thus only occur in the presence of 7 cytochrome c molecules, 7 Apaf-1 proteins and sufficient (d) ATP/ATP .[7] The ATPase domain belongs to the AAA+ family of ATPases; this family is known for its ability to link to other ATPase domains and form hexa- or heptamers.[16] The apoptosome is then considered active when there are seven Apaf-1 molecules arranged in a wheel structure, oriented such that the NB-ARC domains rest in the centre.[1][16]
Active Apoptosome Action
This functional apoptosome then can provide a platform activation of caspase 9.[1][16] Caspase 9 exists as a zymogen in the cytosol and is thought to be found at 20 nM in cells.[16] Though it is known that the zymogen does not need to be cleaved in order to become active,[16] the activity of procaspase-9 may increase significantly once cleaved.[13] The first hypothesis is that the apoptosome provides a location for the dimerization of two caspase 9 molecules before cleavage; this hypothesis was favoured by Reidl & Salvasen in 2007. The second is that cleavage takes place while caspase 9 is still in its monomeric form.[13][16] In each case, caspase 9 activation leads to the activation of a full caspase cascade and subsequent cell death. It has been suggested that the evolutionary reason for the multimeric protein complex activating the caspase cascade is to ensure trace amounts of cytochrome c do not accidentally cause apoptosis.[7]
Research Areas
What happens when mutations occur?
While apoptosis is required for natural body function, mutations of the apoptosome pathway cause catastrophic effects and changes in the body. Mutations of the cell pathway can either promote cell death or disallow cell death creating a huge amount of disease in the body. Mutated apoptosis pathways causing disease are plentiful and have a wide range from cancer, due to lack of apoptosome activity, Alzheimer's disease due to too much apoptosome activity, and many other neurodegenerative diseases such as Parkinson's disease and Huntington's disease.[18] Neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's are all age-related diseases and involve increased apoptosis where cells die that are still able to function or that contribute to support function of tissue. Apaf-1-ALT is an Apaf-1 mutant found in prostate cancer, which does not have residues 339-1248. Recent structural studies of apoptosome prove that Apaf-1-ALT cannot form apoptosome as it misses key structural components for assembly.[1]
Repression of Apoptosis causing cancer
Genetic and biochemical abnormalities within a cell normally trigger programmed cell death to rid the body of irregular cell function and development; however, cancer cells have acquired mutations that allow them to repress apoptosis and survive. Chemotherapies like ionizing radiation have been developed to activate these repressed PCD pathways by hyper-stimulation to promote normal PCD.[19]
P53 mutations in Apoptosis
P53 functions as a tumor suppressor that is involved in preventing cancer and occurs naturally in apoptotic pathways. P53 causes cells to enter apoptosis and disrupt further cell division therefore preventing that cell from becoming cancerous (16). In the majority of cancers it is the p53 pathway that has become mutated resulting in lack of ability to terminate dysfunctional cells. P53 function can also be responsible for a limited life span where mutations of the p53 gene causes expression of dominant-negative forms producing long lived animals. For example in an experiment using C. elegans, the increased life span of p53 mutants was found to depend on increased autophagy.[19] In another experiment using Drosophilia the p53 mutation had both positive and negative effects on the adult life span, which concluded a link between sexual differentiation, PCD and aging.[19] Determining how p53 are affecting life span will be an important area for future research.
Targeting the Apoptosome for Cancer therapy
The inhibition of apoptosis is one of the key features of cancer so finding ways to manipulate and overcome this inhibition to form the apoptosome and activate caspases are important in the development of new cancer treatments.[20] The ability to directly cause apoptosome activation is valuable in cancer therapies because the infected cancerous genes are unable to be destroyed causing a continuation of the cancer to form. By activating the apoptosome by an outside stimulus apoptosis can occur and get rid of the mutated cells. Numerous approaches to achieve this are currently being pursued including recombinant biomolecules, antisense strategies, gene therapy and classic organic combinatorial chemistry to target specific apoptotic regulators in the approach to correct excessive or deficient cell death in human diseases.[18]
In general the up regulation of anti-apoptotic proteins leads to the prevention of apoptosis which can be solved by inhibitors and the down regulation of anti-apoptotic proteins leads to the induction of apoptosis which is reversed by activators that are able to bind and modify their activity. An important target molecule in apoptosis based therapies is Bcl-2 for drug design.[18] Bcl-2 was the first oncogene found to cause cancer-inhibiting apoptosis. It is over expressed in tumors and is resistant to chemotherapy.[18] Scientists have found that binding depressors to Bcl-2 anti-apoptotic proteins inhibits them and leaves direct activators free to interact with Bax and Bak.[18]
Another targeted molecule for cancer therapy involves the caspase family and their regulators. The inhibition of caspase activity blocks cell death in human disease including neurodegenerative disorders, stroke, heart attack and liver injury. Therefore, caspase inhibitors are a promising pharmacological tool providing treatments for stroke and other human diseases. There are several caspase inhibitors that are currently in the preclinical stage that have shown promising evidence of reversing effects of some neurodegenerative diseases. In a recent study researchers developed a reversible caspase-3 inhibitor called M-826 and tested it in a mouse model where it blocked brain tissue damage. Furthermore, it was tested on a mouse with Huntington's disease and the inhibitor prevented striated neuron death revealing promising effects for further study of this caspase inhibitor.[18]
Apoptosome complex has revealed new potential targets for molecular therapy
The Apaf1/caspase-9 apoptosome formation is a crucial event in the apoptotic cascade. The identification of new potential drugs that prevent or stabilize the formation of active apoptosome complex is the ideal strategy for the treatment of disease characterized by excessive or insufficient apoptosis.[18] Recently taurine has been found to prevent ischemia-induced apoptosis in cardiomyocytes through its ability to inhibit Apaf1/caspase-9 apoptosome formation without preventing mitochondrial dysfunction. The possible mechanism by which taurine inhibits the apoptosome formation was identified as being capable of reducing the expression of caspase-9, a fundamental component of apoptosome. However, there are studies that show Aparf1 and caspase-9 have independent roles other than the apoptosome so altering their levels could alter cell function as well. So despite encouraging experimental data several problems remain unsolved and limit the use of experimental drugs in clinical practice.[18]
The discovery of apoptosome inhibitors will provide a new therapeutical tool for the treatment of apoptosis mediated disease. Of particular importance are those new compounds able to inhibit apoptosome stability and activity, by acting on intracellular protein–protein interactions without altering the transcriptional levels of the apoptosome components.[18] Recent structural studies of apoptosome may provide valuable tools for designing apoptosome-based therapies.[1][13]
See also
References
- Yuan S, Yu X, Topf M, Ludtke SJ, Wang X, Akey CW. "Structure of an apoptosome-procaspase-9 CARD complex." Structure. 2010 May;18(5):571-83.
- Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW (February 2002). "Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation". Molecular Cell. 9 (2): 423–32. doi:10.1016/s1097-2765(02)00442-2. PMID 11864614.
- T. F. Reubold; S. Wohlgemuth; S. Eschenburg (2011). "Crystal structure of full-length Apaf-1: how the death signal is relayed in the mitochondrial pathway of apoptosis". Structure. 19 (8): 1074–1083. doi:10.1016/j.str.2011.05.013. PMID 21827944.
- Tsujimoto Y (November 1998). "Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria?". Genes to Cells. 3 (11): 697–707. doi:10.1046/j.1365-2443.1998.00223.x. PMID 9990505.
- Pan, G.; O'Rourke, K.; Dixit, V. M. (1998). "Caspase-9, Bcl-XL, and Apaf-1 Form a Ternary Complex". Journal of Biological Chemistry. 273 (10): 5841–845. doi:10.1074/jbc.273.10.5841. PMID 9488720.
- Hu, Ding; Spencer; Nunez (1998). "WD-40 Repeat Region Regulates Apaf-1 Self-association and Procaspase-9 Activation". Journal of Biological Chemistry. 273 (50): 33489–3494. doi:10.1074/jbc.273.50.33489. PMID 9837928.
- Zou H, Li Y, Liu X, Wang X (April 1999). "An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9". The Journal of Biological Chemistry. 274 (17): 11549–56. doi:10.1074/jbc.274.17.11549. PMID 10206961.
- Jia L, Srinivasula SM, Liu FT, Newland AC, Fernandes-Alnemri T, Alnemri ES, Kelsey SM (July 2001). "Apaf-1 protein deficiency confers resistance to cytochrome c-dependent apoptosis in human leukemic cells". Blood. 98 (2): 414–21. doi:10.1182/blood.v98.2.414. PMID 11435311.
- Bitzer M, Armeanu S, Prinz F, Ungerechts G, Wybranietz W, Spiegel M, Bernlöhr C, Cecconi F, Gregor M, Neubert WJ, Schulze-Osthoff K, Lauer UM (August 2002). "Caspase-8 and Apaf-1-independent caspase-9 activation in Sendai virus-infected cells". The Journal of Biological Chemistry. 277 (33): 29817–9824. doi:10.1074/jbc.M111898200. PMID 12021264.
- Wolf BB, Schuler M, Li W, Eggers-Sedlet B, Lee W, Tailor P, Fitzgerald P, Mills GB, Green DR (September 2001). "Defective cytochrome c-dependent caspase activation in ovarian cancer cell lines due to diminished or absent apoptotic protease activating factor-1 activity". The Journal of Biological Chemistry. 276 (36): 34244–51. doi:10.1074/jbc.M011778200. PMID 11429402.
- Belmokhtar CA, Hillion J, Dudognon C, Fiorentino S, Flexor M, Lanotte M, Ségal-Bendirdjian E (August 2003). "Apoptosome-independent pathway for apoptosis. Biochemical analysis of APAF-1 defects and biological outcomes". The Journal of Biological Chemistry. 278 (32): 29571–80. doi:10.1074/jbc.M302924200. PMID 12773531.
- Ceconi F, Ferraro E, Fuoco C, Strappazzon F (2010). "Apoptosome Structure and Regulation". Apoptosome: 27–39. doi:10.1007/978-90-481-3415-1_2. ISBN 978-90-481-3414-4.
- Yuan S, Yu X, Asara JM, Heuser JE, Ludtke SJ and Akey CW. "The Holo-Apoptosome: Activation of Procaspase-9 and Interactions with Caspase-3." Structure. 2011 August;19(8):1084-1096.
- Yuan S, Yu X, Topf M, Dorstyn L, Kumar S, Ludtke SJ and Akey CW. "Structure of the Drosophila Apoptosome at 6.9 Å Resolution." Structure. 2011 January;19(1):128-140.
- S. Qi; Y. Pang; Q. Hu; Q. Liu; H. Li; Y. Zhou; T. He; Q. Liang; Y. Liu; X. Yuan; G. Luo; H. Li; J. Wang; N. Yan; Y. Shi (2010). "Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4". Cell. 141 (3): 446–457. doi:10.1016/j.cell.2010.03.017. PMID 20434985.
- Riedl SJ, Salvesen GS (May 2007). "The apoptosome: signalling platform of cell death". Nature Reviews. Molecular Cell Biology. 8 (5): 405–13. doi:10.1038/nrm2153. PMID 17377525.
- Diamond & McCabe (2007). The mitochondrion and plant programmed cell death. In: Logan DC (Ed.), Plant mitochondria. Annual Plant Review 2007, 31:308–334.
- D'Amelio M, Tino E, Cecconi F (April 2008). "The apoptosome: emerging insights and new potential targets for drug design". Pharmaceutical Research. 25 (4): 740–51. doi:10.1007/s11095-007-9396-z. PMC 2279152. PMID 17674158.
- Shen J, Tower J (December 2009). "Programmed cell death and apoptosis in aging and life span regulation". Discovery Medicine. 8 (43): 223–6. PMID 20040274.
- Fischer U, Janssen K, Schulze-Osthoff K (2007). "Cutting-edge apoptosis-based therapeutics: a panacea for cancer?". BioDrugs : Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy. 21 (5): 273–97. doi:10.2165/00063030-200721050-00001. PMID 17896835.