Chorismate mutase
In enzymology, chorismate mutase (EC 5.4.99.5) is an enzyme that catalyzes the chemical reaction for the conversion of chorismate to prephenate in the pathway to the production of phenylalanine and tyrosine, also known as the shikimate pathway. Hence, this enzyme has one substrate, chorismate, and one product, prephenate. Chorismate mutase is found at a branch point in the pathway. The enzyme channels the substrate, chorismate to the biosynthesis of tyrosine and phenylalanine and away from tryptophan.[1] Its role in maintaining the balance of these aromatic amino acids in the cell is vital.[2] This is the single known example of a naturally occurring enzyme catalyzing a pericyclic reaction.[2][nb 1] Chorismate mutase is only found in fungi, bacteria, and higher plants. Some varieties of this protein may use the morpheein model of allosteric regulation.[4]
| Chorismate mutase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
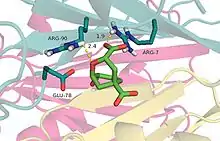 Crystal structure of chorismate mutase with a transition state analogue bound | |||||||||
| Identifiers | |||||||||
| EC number | 5.4.99.5 | ||||||||
| CAS number | 9068-30-8 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
Protein family

This enzyme belongs to the family of isomerases, specifically those intramolecular transferases that transfer functional groups. The systematic name of this enzyme class is chorismate pyruvatemutase. Chorismate mutase, also known as hydroxyphenylpyruvate synthase, participates in phenylalanine, tyrosine and tryptophan biosynthesis.[1] The structures of chorismate mutases vary in different organisms, but the majority belong to the AroQ family and are characterized by an intertwined homodimer of 3-helical subunits. Most chorismate mutases in this family look similar to that of Escherichia coli. For example, the secondary structure of the chorismate mutase of yeast is very similar to that of E. coli. Chorimate mutase in the AroQ family are more common in nature and are widely distributed among the prokaryotes.[1] For optimal function, they usually have to be accompanied by another enzyme such as prephanate dehydrogenase. These chorismate mutases are typically bifunctional enzymes, meaning they contain two catalytic capacities in the same polypeptide chain.[1] However, the chorismate mutase of eukaryotic organisms are more commonly monofunctional. There are organisms such as Bacillus subtilis whose chorismate mutase have a completely different structure and are monofunctional. These enzymes belong to the AroH family and are characterized by a trimeric α/β barrel topology.[5]
Mechanism of catalysis
The conversion of chorismate to prephenate is the first committed step in the pathway to the production of the aromatic amino acids: tyrosine and phenylalanine. The presence of chorismate mutase increases the rate of the reaction a million fold.[6] In the absence of enzyme catalysis this mechanism proceeds as a concerted, but asynchronous step and is an exergonic process. The mechanism for this transformation is formally a Claisen rearrangement, supported by the kinetic and isotopic data reported by Knowles, et al[7]
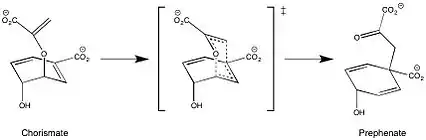
E. coli and Yeast chorismate mutase have a limited sequence homology, but their active sites contain similar residues. The active site of the Yeast chorismate mutase contains Arg16, Arg157, Thr242, Glu246, Glu198, Asn194, and Lys168. The E. coli active site contains the same residues with the exception of these noted exchanges: Asp48 for Asn194, Gln88 for Glu248, and Ser84 for Thr242. In the enzyme active site, interactions between these specific residues and the substrate restrict conformational degrees of freedom, such that the entropy of activation is effectively reduced to zero, and thereby promotes catalysis. As a result, there is no formal intermediate, but rather a pseudo-diaxial chair-like transition state. Evidence for this conformation is provided by an inverse secondary kinetic isotope effect at the carbon directly attached to the hydroxyl group.[6] This seemingly unfavorable arrangement is achieved through a series of electrostatic interactions, which rotate the extended chain of chorismate into the conformation required for this concerted mechanism.
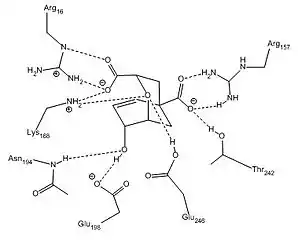
An additional stabilizing factor in this enzyme-substrate complex is hydrogen bonding between the lone pair of the oxygen in the vinyl ether system and hydrogen bond donor residues. Not only does this stabilize the complex, but disruption of resonance within the vinyl ether destabilizes the ground state and reduces the energy barrier for this transformation. An alternative view is that electrostatic stabilization of the polarized transition state is of great importance in this reaction. In the chorismate mutase active site, the transition-state analog is stabilized by 12 electrostatic and hydrogen-bonding interactions.[8] This is shown in mutants of the native enzyme in which Arg90 is replaced with citrulline to demonstrate the importance of hydrogen bonding to stabilize the transition state.[9] Other work using chorismate mutase from Bacillus subtilis showed evidence that when a cation was aptly placed in the active site, the electrostatic interactions between it and the negatively charged transition state promoted catalysis.[2]
Additional studies have been done in order to support the relevance of a near attack conformer (NAC) in the reaction catalyzed by chorismate mutase. This NAC is the reactive conformation of the ground state that is directly converted to the transition state in the enzyme. Using thermodynamic integration (TI) methods, the standard free energies (ΔGN°) for NAC formation were calculated in six different environments. The data obtained suggests that effective catalysis is derived from stabilization of both the NAC and transition state.[10] However, other experimental evidence supports that the NAC effect observed is simply a result of electrostatic transition state stabilization.[11][12]
Overall, there have been extensive studies on the exact mechanism of this reaction. However, the relative contribution of conformational constraint of the flexible substrate, specific hydrogen bonding to the transition state, and electrostatic interactions to the observed rate enhancement is still under discussion.
Notes
- Dimethylallyltryptophan synthase has been proposed to catalyze a Cope rearrangement, but this has yet to be proven definitively[3]
References
- Qamra R, Prakash P, Aruna B, Hasnain SE, Mande SC (June 2006). "The 2.15 A crystal structure of Mycobacterium tuberculosis chorismate mutase reveals an unexpected gene duplication and suggests a role in host-pathogen interactions". Biochemistry. 45 (23): 6997–7005. doi:10.1021/bi0606445. PMID 16752890.
- Kast P, Grisostomi C, Chen IA, Li S, Krengel U, Xue Y, Hilvert D (November 2000). "A strategically positioned cation is crucial for efficient catalysis by chorismate mutase". The Journal of Biological Chemistry. 275 (47): 36832–8. doi:10.1074/jbc.M006351200. PMID 10960481.
- Luk LY, Qian Q, Tanner ME (August 2011). "A cope rearrangement in the reaction catalyzed by dimethylallyltryptophan synthase?". Journal of the American Chemical Society. 133 (32): 12342–5. doi:10.1021/ja2034969. PMID 21766851.
- Selwood T, Jaffe EK (March 2012). "Dynamic dissociating homo-oligomers and the control of protein function". Archives of Biochemistry and Biophysics. 519 (2): 131–43. doi:10.1016/j.abb.2011.11.020. PMC 3298769. PMID 22182754.
- Babu M (1999). "Annotation of Chorismate Mutase from the Mycobacterium tuberculosis and the Mycobacterium leprae genome" (PDF). Undergraduate Thesis for the Center of Biotechnology.
- Lee AY, Stewart JD, Clardy J, Ganem B (April 1995). "New insight into the catalytic mechanism of chorismate mutases from structural studies". Chemistry & Biology. 2 (4): 195–203. doi:10.1016/1074-5521(95)90269-4. PMID 9383421.
- Gray JV, Knowles JR (August 1994). "Monofunctional chorismate mutase from Bacillus subtilis: FTIR studies and the mechanism of action of the enzyme". Biochemistry. 33 (33): 9953–9. doi:10.1021/bi00199a018. PMID 8061004.
- Grisham C (2017). Biochemistry 6th Edition. United States of America: Brooks/Cole - Cengage Learning. p. 505. ISBN 978-1133106296.
- Kienhöfer A, Kast P, Hilvert D (March 2003). "Selective stabilization of the chorismate mutase transition state by a positively charged hydrogen bond donor". Journal of the American Chemical Society. 125 (11): 3206–7. doi:10.1021/ja0341992. PMID 12630863.
- Hur S, Bruice TC (October 2003). "The near attack conformation approach to the study of the chorismate to prephenate reaction". Proceedings of the National Academy of Sciences of the United States of America. 100 (21): 12015–20. doi:10.1073/pnas.1534873100. PMC 218705. PMID 14523243.
- Strajbl M, Shurki A, Kato M, Warshel A (August 2003). "Apparent NAC effect in chorismate mutase reflects electrostatic transition state stabilization". Journal of the American Chemical Society. 125 (34): 10228–37. doi:10.1021/ja0356481. PMID 12926945.
- Burschowsky D, van Eerde A, Ökvist M, Kienhöfer A, Kast P, Hilvert D, Krengel U (December 2014). "Electrostatic transition state stabilization rather than reactant destabilization provides the chemical basis for efficient chorismate mutase catalysis". Proceedings of the National Academy of Sciences of the United States of America. 111 (49): 17516–21. Bibcode:2014PNAS..11117516B. doi:10.1073/pnas.1408512111. PMC 4267393. PMID 25422475.