Chromosome conformation capture
Chromosome conformation capture techniques (often abbreviated to 3C technologies or 3C-based methods[1]) are a set of molecular biology methods used to analyze the spatial organization of chromatin in a cell. These methods quantify the number of interactions between genomic loci that are nearby in 3-D space, but may be separated by many nucleotides in the linear genome.[2] Such interactions may result from biological functions, such as promoter-enhancer interactions, or from random polymer looping, where undirected physical motion of chromatin causes loci to collide.[3] Interaction frequencies may be analyzed directly,[4] or they may be converted to distances and used to reconstruct 3-D structures.[5]
.svg.png.webp)
The chief difference between 3C-based methods is their scope. For example, when using PCR to detect interaction in a 3C experiment, the interactions between two specific fragments are quantified. In contrast, Hi-C quantifies interactions between all possible pairs of fragments simultaneously. Deep sequencing of material produced by 3C also produces genome-wide interactions maps.
History
Historically, microscopy was the primary method of investigating nuclear organization,[6] which can be dated back to 1590.[7]
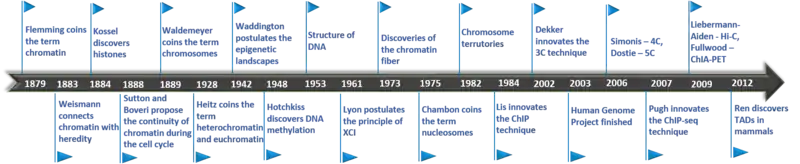
- In 1879, Walther Flemming coined the term chromatin.[8]
- In 1883, August Weismann connected chromatin with heredity.
- In 1884, Albrecht Kossel discovered histones.
- In 1888, Sutton and Boveri proposed the theory of continuity of chromatin during the cell cycle [9]
- In 1889, Wilhelm von Waldemeyer created the term "chromosome".[10]
- In 1928, Emil Heitz coined the term Heterochromatin and Euchromatin.[11]
- In 1942, Conrad Waddington postulated the epigenetic landscapes.[12]
- In 1948, Rollin Hotchkiss discovered DNA methylation.[13]
- In 1953, Watson and Crick discovered the double helix structure of DNA.[14]
- In 1961, Mary Lyon postulated the principle of X-inactivation.
- In 1973/1974, chromatin fiber was discovered.[12]
- In 1975, Pierre Chambon coined the term nucleosomes.[12]
- In 1982, Chromosome territories were discovered.[15]
- In 1984, John T. Lis innovated the Chromatin immunoprecipitation technique.
- In 1993, the Nuclear Ligation Assay was published, a method that could determine circularization frequencies of DNA in solution. This assay was used to show that estrogen induces an interaction between the prolactin gene promoter and a nearby enhancer.[16]
- In 2002, Job Dekker introduced the new idea that dense matrices of interaction frequencies between loci could be used to infer the spatial organization of genomes. This idea was the basis for his development of the chromosome conformation capture (3C) assay, published in 2002 by Job Dekker and colleagues in the Kleckner lab at Harvard University.[17][18]
- In 2003, the Human Genome Project was finished.
- In 2006, Marieke Simonis invented 4C,[19] Dostie, in the Dekker lab, invented 5C.[20]
- In 2007, B. Franklin Pugh innovated ChIP-seq technique.[21]
- In 2009, Lieberman-Aiden and Job Dekker invented Hi-C,[22] Melissa J. Fullwood and Yijun Ruan invented ChIA-PET.[23]
- In 2012, The Ren group, and the groups led by Edith Heard and Job Dekker discovered Topologically Associating Domains (TADs) in mammals.[24][25]
- In 2013, Takashi Nagano and Peter Fraser introduced in-nuclei ligation for Hi-C and single-cell Hi-C.[26]
Experimental methods
All 3C methods start with a similar set of steps, performed on a sample of cells.
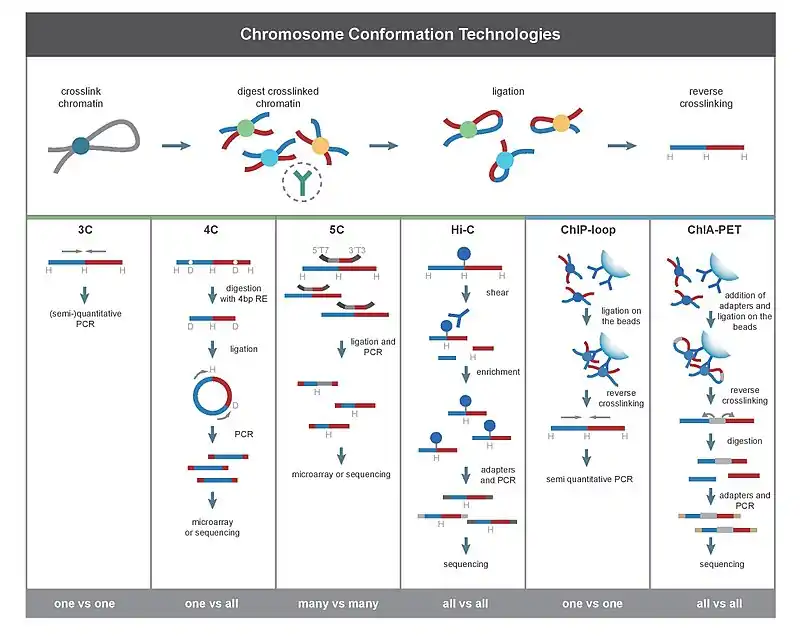
First, the cell genomes are cross-linked with formaldehyde,[27] which introduces bonds that "freeze" interactions between genomic loci. Treatment of cells with 1-3% formaldehyde, for 10-30min at room temperature is most common, however, standardization for preventing high protein-DNA cross linking is necessary, as this may negatively affect the efficiency of restriction digestion in the subsequent step.[28] The genome is then cut into fragments with a restriction endonuclease. The size of restriction fragments determines the resolution of interaction mapping. Restriction enzymes (REs) that make cuts on 6bp recognition sequences, such as EcoR1 or HindIII, are used for this purpose, as they cut the genome once every 4000bp, giving ~ 1 million fragments in the human genome.[28][29] For more precise interaction mapping, a 4bp recognizing RE may also be used. The next step is, proximity based ligation. This takes place at low DNA concentrations or within intact, permeabilized nuclei[26] in the presence of T4 DNA ligase,[30] such that ligation between cross-linked interacting fragments is favored over ligation between fragments that are not cross-linked. Subsequently, interacting loci are quantified by amplifying ligated junctions by PCR methods.[28][30]
3C (one-vs-one)
The chromosome conformation capture (3C) experiment quantifies interactions between a single pair of genomic loci. For example, 3C can be used to test a candidate promoter-enhancer interaction. Ligated fragments are detected using PCR with known primers.[2][17] That is why this technique requires the prior knowledge of the interacting regions.
4C (one-vs-all)
Chromosome conformation capture-on-chip (4C) captures interactions between one locus and all other genomic loci. It involves a second ligation step, to create self-circularized DNA fragments, which are used to perform inverse PCR. Inverse PCR allows the known sequence to be used to amplify the unknown sequence ligated to it.[31][2][19] In contrast to 3C and 5C, the 4C technique does not require the prior knowledge of both interacting chromosomal regions. Results obtained using 4C are highly reproducible with most of the interactions that are detected between regions proximal to one another. On a single microarray, approximately a million interactions can be analyzed.
5C (many-vs-many)
Chromosome conformation capture carbon copy (5C) detects interactions between all restriction fragments within a given region, with this region's size typically no greater than a megabase.[2][20] This is done by ligating universal primers to all fragments. However, 5C has relatively low coverage. The 5C technique overcomes the junctional problems at the intramolecular ligation step and is useful for constructing complex interactions of specific loci of interest. This approach is unsuitable for conducting genome-wide complex interactions since that will require millions of 5C primers to be used.
Hi-C (all-vs-all)
Hi-C uses high-throughput sequencing to find the nucleotide sequence of fragments[2][22] and uses paired end sequencing, which retrieves a short sequence from each end of each ligated fragment. As such, for a given ligated fragment, the two sequences obtained should represent two different restriction fragments that were ligated together in the proximity based ligation step. The pair of sequences are individually aligned to the genome, thus determining the fragments involved in that ligation event. Hence, all possible pairwise interactions between fragments are tested.
Sequence capture-based methods
A number of methods use oligonucleotide capture to enrich 3C and Hi-C libraries for specific loci of interest.[32][33] These methods include Capture-C,[34] NG Capture-C,[35] Capture-3C,[34] HiCap,[32][36] and Capture Hi-C.[37] These methods are able to produce higher resolution and sensitivity than 4C based methods.[38]
Single-cell methods
Single-cell adaptations of these methods, such as ChIP-seq and Hi-C can be used to investigate the interactions occurring in individual cells.[39][40]
ChIP-loop
ChIP-loop combines 3C with ChIP-seq to detect interactions between two loci of interest mediated by a protein of interest.[2][41] The ChIP-loop may be useful in identifying long-range cis-interactions and trans interaction mediated through proteins since frequent DNA collisions will not occur.
Biological impact
3C methods have led to a number of biological insights, including the discovery of new structural features of chromosomes, the cataloguing of chromatin loops, and increased understanding of transcriptional regulation mechanisms (the disruption of which can lead to disease).[6]
3C methods have demonstrated the importance of spatial proximity of regulatory elements to the genes that they regulate. For example, in tissues that express globin genes, the β-globin locus control region forms a loop with these genes. This loop is not found in tissues where the gene is not expressed.[43] This technology has further aided the genetic and epigenetic study of chromosomes both in model organisms and in humans.
These methods have revealed large-scale organization of the genome into topologically associating domains (TADs), which correlate with epigenetic markers. Some TADs are transcriptionally active, while others are repressed.[44] Many TADs have been found in D. melanogaster, mouse and human.[45] Moreover, CTCF and cohesin play important roles in determining TADs and enhancer-promoter interactions. The result shows that the orientation of CTCF binding motifs in an enhancer-promoter loop should be facing to each other in order for the enhancer to find its correct target.[46]
Human disease
There are several diseases caused by defects in promoter-enhancer interactions, which is reviewed in this paper.[47]
Beta thalassemia is a certain type of blood disorders caused by a deletion of LCR enhancer element.[48][49]
Holoprosencephaly is cephalic disorder caused by a mutation in the SBE2 enhancer element, which in turn weakened the production of SHH gene.[50]
PPD2 (polydactyly of a triphalangeal thumb) is caused by a mutation of ZRS enhancer, which in turn strengthened the production of SHH gene.[51][52]
Adenocarcinoma of the lung can be caused by a duplication of enhancer element for MYC gene.[53]
T-cell acute lymphoblastic leukemia is caused by an introduction of a new enhancer.[54]
Data analysis
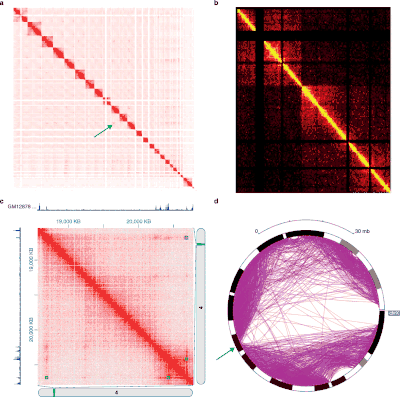
The different 3C-style experiments produce data with very different structures and statistical properties. As such, specific analysis packages exist for each experiment type.[33]
Hi-C data is often used to analyze genome-wide chromatin organization, such as topologically associating domains (TADs), linearly contiguous regions of the genome that are associated in 3-D space.[44] Several algorithms have been developed to identify TADs from Hi-C data.[4][59]
Hi-C and its subsequent analyses are evolving. Fit-Hi-C [3] is a method based on a discrete binning approach with modifications of adding distance of interaction (initial spline fitting, aka spline-1) and refining the null model (spline-2). The result of Fit-Hi-C is a list of pairwise intra-chromosomal interactions with their p-values and q-values.[58]
The 3-D organization of the genome can also be analyzed via eigendecomposition of the contact matrix. Each eigenvector corresponds to a set of loci, which are not necessarily linearly contiguous, that share structural features.[60]
A significant confounding factor in 3C technologies is the frequent non-specific interactions between genomic loci that occur due to random polymer behavior. An interaction between two loci must be confirmed as specific through statistical significance testing.[3]
Normalization of Hi-C contact map
There are two major ways of normalizing raw Hi-C contact heat maps. The first way is to assume equal visibility, meaning there is an equal chance for each chromosomal position to have an interaction. Therefore, the true signal of a Hi-C contact map should be a balanced matrix (Balanced matrix has constant row sums and column sums). An example of algorithms that assumes equal visibility is Sinkhorn-Knopp algorithm, which scales the raw Hi-C contact map into a balanced matrix.
The other way is to assume there is a bias associated with each chromosomal position. The contact map value at each coordinate will be the true signal at that position times bias associated with the two contact positions. An example of algorithms that aim to solve this model of bias is iterative correction, which iteratively regressed out row and column bias from the raw Hi-C contact map. There are a number of software tools available for analysis of Hi-C data.[61]
DNA motif analysis
DNA motifs are specific short DNA sequences, often 8-20 nucleotides in length[62] which are statistically overrepresented in a set of sequences with a common biological function. Currently, regulatory motifs on the long-range chromatin interactions have not been studied extensively. Several studies have focused on elucidating the impact of DNA motifs in promoter-enhancer interactions.
Bailey et al. has identified that ZNF143 motif in the promoter regions provides sequence specificity for promoter-enhancer interactions.[63] Mutation of ZNF143 motif decreased the frequency of promoter-enhancer interactions suggesting that ZNF143 is a novel chromatin-looping factor.
For genome-scale motif analysis, in 2016, Wong et al. reported a list of 19,491 DNA motif pairs for K562 cell line on the promoter-enhancer interactions.[64] As a result, they proposed that motif pairing multiplicity (number of motifs that are paired with a given motif) is linked to interaction distance and regulatory region type. In the next year, Wong published another article reporting 18,879 motif pairs in 6 human cell lines.[65] A novel contribution of this work is MotifHyades, a motif discovery tool that can be directly applied to paired sequences.
Cancer genome analysis
The 3C-based techniques can provide insights into the chromosomal rearrangements in the cancer genomes.[66] Moreover, they can show changes of spatial proximity for regulatory elements and their target genes, which bring deeper understanding of the structural and functional basis of the genome.[67]
References
- de Wit E, de Laat W (January 2012). "A decade of 3C technologies: insights into nuclear organization". Genes & Development. 26 (1): 11–24. doi:10.1101/gad.179804.111. PMC 3258961. PMID 22215806.
- Hakim O, Misteli T (March 2012). "SnapShot: Chromosome confirmation capture". Cell. 148 (5): 1068.e1–2. doi:10.1016/j.cell.2012.02.019. PMC 6374129. PMID 22385969.
- Ay F, Bailey TL, Noble WS (June 2014). "Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts". Genome Research. 24 (6): 999–1011. doi:10.1101/gr.160374.113. PMC 4032863. PMID 24501021.
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL (December 2014). "A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping". Cell. 159 (7): 1665–80. doi:10.1016/j.cell.2014.11.021. PMC 5635824. PMID 25497547.
- Varoquaux N, Ay F, Noble WS, Vert JP (June 2014). "A statistical approach for inferring the 3D structure of the genome". Bioinformatics. 30 (12): i26–33. doi:10.1093/bioinformatics/btu268. PMC 4229903. PMID 24931992.
- Denker A, de Laat W (June 2016). "The second decade of 3C technologies: detailed insights into nuclear organization". Genes & Development. 30 (12): 1357–82. doi:10.1101/gad.281964.116. PMC 4926860. PMID 27340173.
- "Who invented the microscope? A complete Microscope History". Vision Engineering Ltd. Archived from the original on 22 April 2018.
- "Photography by Benjamin Saur Tübingen Walther Flemming a German Physician". Course Hero, Inc.
- Martins LA (1999). "Did Sutton and Boveri propose the so-called Sutton-Boveri chromosome hypothesis?". Genet. Mol. Biol. 22 (2): 261–272. doi:10.1590/S1415-47571999000200022.
- "Genes and genetics: The language of scientific discovery". Oxford English Dictionary. Oxford University Press. 2012-08-16.
- Harris M (2015-02-05). "Heterochromatin and euchromatin mains".
- Deichmann U (August 2016). "Epigenetics: The origins and evolution of a fashionable topic". Developmental Biology. 416 (1): 249–254. doi:10.1016/j.ydbio.2016.06.005. PMID 27291929.
- Lu H, Liu X, Deng Y, Qing H (December 2013). "DNA methylation, a hand behind neurodegenerative diseases". Frontiers in Aging Neuroscience. 5: 85. doi:10.3389/fnagi.2013.00085. PMC 3851782. PMID 24367332.
- "The Francis Crick Papers: The Discovery of the Double Helix, 1951–1953".
- Cremer T, Cremer M (March 2010). "Chromosome territories". Cold Spring Harbor Perspectives in Biology. 2 (3): a003889. doi:10.1101/cshperspect.a003889. PMC 2829961. PMID 20300217.
- Cullen KE, Kladde MP, Seyfred MA (July 1993). "Interaction between transcription regulatory regions of prolactin chromatin". Science. 261 (5118): 203–6. Bibcode:1993Sci...261..203C. doi:10.1126/science.8327891. PMID 8327891.
- Dekker J, Rippe K, Dekker M, Kleckner N (February 2002). "Capturing chromosome conformation". Science. 295 (5558): 1306–11. Bibcode:2002Sci...295.1306D. doi:10.1126/science.1067799. PMID 11847345. S2CID 3561891.
- Osborne CS, Ewels PA, Young AN (January 2011). "Meet the neighbours: tools to dissect nuclear structure and function". Briefings in Functional Genomics. 10 (1): 11–7. doi:10.1093/bfgp/elq034. PMC 3080762. PMID 21258046.
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W (November 2006). "Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C)". Nature Genetics. 38 (11): 1348–54. doi:10.1038/ng1896. PMID 17033623. S2CID 22787572.
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, et al. (October 2006). "Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements". Genome Research. 16 (10): 1299–309. doi:10.1101/gr.5571506. PMC 1581439. PMID 16954542.
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF (March 2007). "Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome". Nature. 446 (7135): 572–6. Bibcode:2007Natur.446..572A. doi:10.1038/nature05632. PMID 17392789. S2CID 4416890.
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. (October 2009). "Comprehensive mapping of long-range interactions reveals folding principles of the human genome". Science. 326 (5950): 289–93. Bibcode:2009Sci...326..289L. doi:10.1126/science.1181369. PMC 2858594. PMID 19815776.
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, et al. (November 2009). "An oestrogen-receptor-alpha-bound human chromatin interactome". Nature. 462 (7269): 58–64. Bibcode:2009Natur.462...58F. doi:10.1038/nature08497. PMC 2774924. PMID 19890323.
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (April 2012). "Topological domains in mammalian genomes identified by analysis of chromatin interactions". Nature. 485 (7398): 376–80. Bibcode:2012Natur.485..376D. doi:10.1038/nature11082. PMC 3356448. PMID 22495300.
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E (April 2012). "Spatial partitioning of the regulatory landscape of the X-inactivation centre". Nature. 485 (7398): 381–5. Bibcode:2012Natur.485..381N. doi:10.1038/nature11049. PMC 3555144. PMID 22495304.
- Nagano, Takashi; Lubling, Yaniv; Stevens, Tim J.; Schoenfelder, Stefan; Yaffe, Eitan; Dean, Wendy; Laue, Ernest D.; Tanay, Amos; Fraser, Peter (October 2013). "Single-cell Hi-C reveals cell-to-cell variability in chromosome structure". Nature. 502 (7469): 59–64. Bibcode:2013Natur.502...59N. doi:10.1038/nature12593. PMC 3869051. PMID 24067610.
- Gavrilov A, Eivazova E, Priozhkova I, Lipinski M, Razin S, Vassetzky Y (2009). "Chromosome conformation capture (from 3C to 5C) and its ChIP-based modification". Chromatin Immunoprecipitation Assays. review. Methods in Molecular Biology. 567. pp. 171–88. doi:10.1007/978-1-60327-414-2_12. ISBN 978-1-60327-413-5. PMID 19588093.
- Naumova N, Smith EM, Zhan Y, Dekker J (November 2012). "Analysis of long-range chromatin interactions using Chromosome Conformation Capture". Methods. 58 (3): 192–203. doi:10.1016/j.ymeth.2012.07.022. PMC 3874837. PMID 22903059.
- Belton JM, Dekker J (June 2015). "Chromosome Conformation Capture (3C) in Budding Yeast". Cold Spring Harbor Protocols. 2015 (6): 580–6. doi:10.1101/pdb.prot085175. PMID 26034304.
- Gavrilov AA, Golov AK, Razin SV (2013-03-26). "Actual ligation frequencies in the chromosome conformation capture procedure". PLOS ONE. 8 (3): e60403. Bibcode:2013PLoSO...860403G. doi:10.1371/journal.pone.0060403. PMC 3608588. PMID 23555968.
- Zhao, Zhihu; Tavoosidana, Gholamreza; Sjolinder, Mikael; Gondor, Anita; Mariano, Piero; Wang, Sha; Kanduri, Chandrasekhar; Lezcano, Magda; Sandhu, Kuljeet Singh; Singh, Umashankar; Pant, Vinod; Tiwari, Vijay; Kurukuti, Sreenivasulu; Ohlsson, Rolf (2006). "Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions". Nature Genetics. 38 (11): 1341–7. doi:10.1038/ng1891. PMID 17033624. S2CID 2660843.
- US patent 10287621
- Schmitt AD, Hu M, Ren B (December 2016). "Genome-wide mapping and analysis of chromosome architecture". Nature Reviews Molecular Cell Biology. 17 (12): 743–755. doi:10.1038/nrm.2016.104. PMC 5763923. PMID 27580841.
- Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, et al. (February 2014). "Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment". Nature Genetics. 46 (2): 205–12. doi:10.1038/ng.2871. PMID 24413732. S2CID 205348099.
- Davies JO, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR, Hughes JR (January 2016). "Multiplexed analysis of chromosome conformation at vastly improved sensitivity". Nature Methods. 13 (1): 74–80. doi:10.1038/nmeth.3664. PMC 4724891. PMID 26595209.
- Sahlén, Pelin; Abdullayev, Ilgar; Ramsköld, Daniel; Matskova, Liudmila; Rilakovic, Nemanja; Lötstedt, Britta; Albert, Thomas J.; Lundeberg, Joakim; Sandberg, Rickard (2015-08-03). "Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution". Genome Biology. 16: 156. doi:10.1186/s13059-015-0727-9. ISSN 1474-760X. PMC 4557751. PMID 26313521.
- Jäger R, Migliorini G, Henrion M, Kandaswamy R, Speedy HE, Heindl A, Whiffin N, Carnicer MJ, Broome L, Dryden N, Nagano T, Schoenfelder S, Enge M, Yuan Y, Taipale J, Fraser P, Fletcher O, Houlston RS (February 2015). "Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci". Nature Communications. 6: 6178. Bibcode:2015NatCo...6.6178J. doi:10.1038/ncomms7178. PMC 4346635. PMID 25695508.
- Davies JO, Oudelaar AM, Higgs DR, Hughes JR (January 2017). "How best to identify chromosomal interactions: a comparison of approaches". Nature Methods. 14 (2): 125–134. doi:10.1038/nmeth.4146. PMID 28139673. S2CID 4136037.
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, et al. (October 2013). "Single-cell Hi-C reveals cell-to-cell variability in chromosome structure". Nature. 502 (7469): 59–64. Bibcode:2013Natur.502...59N. doi:10.1038/nature12593. PMC 3869051. PMID 24067610.
- Schwartzman O, Tanay A (December 2015). "Single-cell epigenomics: techniques and emerging applications". Nature Reviews Genetics. 16 (12): 716–26. doi:10.1038/nrg3980. PMID 26460349. S2CID 10326803.
- Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T (January 2005). "Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome". Nature Genetics. 37 (1): 31–40. doi:10.1038/ng1491. PMID 15608638. S2CID 2884412.
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY (November 2016). "HiChIP: efficient and sensitive analysis of protein-directed genome architecture". Nature Methods. 13 (11): 919–922. doi:10.1038/nmeth.3999. PMC 5501173. PMID 27643841.
- Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W (December 2002). "Looping and interaction between hypersensitive sites in the active beta-globin locus". Molecular Cell. 10 (6): 1453–65. doi:10.1016/S1097-2765(02)00781-5. PMID 12504019.
- Cavalli G, Misteli T (March 2013). "Functional implications of genome topology". Nature Structural & Molecular Biology. 20 (3): 290–9. doi:10.1038/nsmb.2474. PMC 6320674. PMID 23463314.
- Dekker J, Marti-Renom MA, Mirny LA (June 2013). "Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data". Nature Reviews Genetics. 14 (6): 390–403. doi:10.1038/nrg3454. PMC 3874835. PMID 23657480.
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, et al. (August 2015). "CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function". Cell. 162 (4): 900–10. doi:10.1016/j.cell.2015.07.038. PMC 4642453. PMID 26276636.
- Krijger PH, de Laat W (December 2016). "Regulation of disease-associated gene expression in the 3D genome". Nature Reviews Molecular Cell Biology. 17 (12): 771–782. doi:10.1038/nrm.2016.138. PMID 27826147. S2CID 11484886.
- Fritsch EF, Lawn RM, Maniatis T (June 1979). "Characterisation of deletions which affect the expression of fetal globin genes in man". Nature. 279 (5714): 598–603. Bibcode:1979Natur.279..598F. doi:10.1038/279598a0. PMID 450109. S2CID 4243029.
- Van der Ploeg LH, Konings A, Oort M, Roos D, Bernini L, Flavell RA (February 1980). "gamma-beta-Thalassaemia studies showing that deletion of the gamma- and delta-genes influences beta-globin gene expression in man". Nature. 283 (5748): 637–42. Bibcode:1980Natur.283..637V. doi:10.1038/283637a0. PMID 6153459. S2CID 4371542.
- Jeong Y, El-Jaick K, Roessler E, Muenke M, Epstein DJ (February 2006). "A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers". Development. 133 (4): 761–72. doi:10.1242/dev.02239. PMID 16407397.
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, et al. (July 2003). "A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly". Human Molecular Genetics. 12 (14): 1725–35. doi:10.1093/hmg/ddg180. PMID 12837695.
- Wieczorek D, Pawlik B, Li Y, Akarsu NA, Caliebe A, May KJ, et al. (January 2010). "A specific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb". Human Mutation. 31 (1): 81–9. doi:10.1002/humu.21142. PMID 19847792. S2CID 1715146.
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M (February 2016). "Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers". Nature Genetics. 48 (2): 176–82. doi:10.1038/ng.3470. PMC 4857881. PMID 26656844.
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. (December 2014). "Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element". Science. 346 (6215): 1373–7. doi:10.1126/science.1259037. PMC 4720521. PMID 25394790.
- Lajoie BR, van Berkum NL, Sanyal A, Dekker J (October 2009). "My5C: web tools for chromosome conformation capture studies". Nature Methods. 6 (10): 690–1. doi:10.1038/nmeth1009-690. PMC 2859197. PMID 19789528.
- Deng X, Ma W, Ramani V, Hill A, Yang F, Ay F, et al. (August 2015). "Bipartite structure of the inactive mouse X chromosome". Genome Biology. 16 (1): 152. doi:10.1186/s13059-015-0728-8. PMC 4539712. PMID 26248554.
- Zhou X, Lowdon RF, Li D, Lawson HA, Madden PA, Costello JF, Wang T (May 2013). "Exploring long-range genome interactions using the WashU Epigenome Browser". Nature Methods. 10 (5): 375–6. doi:10.1038/nmeth.2440. PMC 3820286. PMID 23629413.
- Yardımcı GG, Noble WS (February 2017). "Software tools for visualizing Hi-C data". Genome Biology. 18 (1): 26. doi:10.1186/s13059-017-1161-y. PMC 5290626. PMID 28159004.
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. (April 2012). "Topological domains in mammalian genomes identified by analysis of chromatin interactions". Nature. 485 (7398): 376–80. Bibcode:2012Natur.485..376D. doi:10.1038/nature11082. PMC 3356448. PMID 22495300.
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, et al. (October 2012). "Iterative correction of Hi-C data reveals hallmarks of chromosome organization". Nature Methods. 9 (10): 999–1003. doi:10.1038/nmeth.2148. PMC 3816492. PMID 22941365.
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA (October 2012). "Iterative correction of Hi-C data reveals hallmarks of chromosome organization". Nature Methods. 9 (10): 999–1003. doi:10.1038/nmeth.2148. PMC 3816492. PMID 22941365.
- Zambelli F, Pesole G, Pavesi G (March 2013). "Motif discovery and transcription factor binding sites before and after the next-generation sequencing era". Briefings in Bioinformatics. 14 (2): 225–37. doi:10.1093/bib/bbs016. PMC 3603212. PMID 22517426.
- Bailey, S. D., Zhang, X., Desai, K., Aid, M., Corradin, O., Cowper-Sal·lari, R., … Lupien, M. (2015). ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nature Communications, 2, 6186. Retrieved from https://doi.org/10.1038/ncomms7186
- K. Wong, Y. Li, and C. Peng, “Identification of coupling DNA motif pairs on long-range chromatin interactions in human,” vol. 32, no. September 2015, pp. 321–324, 2016.
- Ka-Chun Wong; MotifHyades: expectation maximization for de novo DNA motif pair discovery on paired sequences, Bioinformatics, Volume 33, Issue 19, 1 October 2017, Pages 3028–3035, https://doi.org/10.1093/bioinformatics/btx381
- Harewood L, Kishore K, Eldridge MD, Wingett S, Pearson D, Schoenfelder S, Collins VP, Fraser P (June 2017). "Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours". Genome Biology. 18 (1): 125. doi:10.1186/s13059-017-1253-8. PMC 5488307. PMID 28655341.
- Taberlay PC, Achinger-Kawecka J, Lun AT, Buske FA, Sabir K, Gould CM, et al. (June 2016). "Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations". Genome Research. 26 (6): 719–31. doi:10.1101/gr.201517.115. PMC 4889976. PMID 27053337.
Further reading
- Barutcu AR, Fritz AJ, Zaidi SK, van Wijnen AJ, Lian JB, Stein JL, Nickerson JA, Imbalzano AN, Stein GS (January 2016). "C-ing the Genome: A Compendium of Chromosome Conformation Capture Methods to Study Higher-Order Chromatin Organization". Journal of Cellular Physiology. 231 (1): 31–5. doi:10.1002/jcp.25062. PMC 4586368. PMID 26059817.
- Marbouty M, Koszul R (December 2015). "Metagenome Analysis Exploiting High-Throughput Chromosome Conformation Capture (3C) Data". review. Trends in Genetics. 31 (12): 673–682. doi:10.1016/j.tig.2015.10.003. PMC 6831814. PMID 26608779.
- Dekker J (25 November 2014). "Two ways to fold the genome during the cell cycle: insights obtained with chromosome conformation capture". Epigenetics & Chromatin. 7 (1): 25. doi:10.1186/1756-8935-7-25. PMC 4247682. PMID 25435919.
- O'Sullivan JM, Hendy MD, Pichugina T, Wake GC, Langowski J (September–October 2013). "The statistical-mechanics of chromosome conformation capture". Nucleus. 4 (5): 390–8. doi:10.4161/nucl.26513. PMC 3899129. PMID 24051548.
- Umbarger MA (November 2012). "Chromosome conformation capture assays in bacteria". review. Methods. 58 (3): 212–20. doi:10.1016/j.ymeth.2012.06.017. PMID 22776362.
- Parelho V, Merkenschlager M (September 2005). "Gene expression: growing up together may help genes go their separate ways". news and commentary. European Journal of Human Genetics. 13 (9): 993–4. doi:10.1038/sj.ejhg.5201464. PMID 15999115. S2CID 29714576.
- Marvin M, Tan-Wong SM (2016-04-23). "Chromosome conformation capture" (commercial method). Abcam PLC. Retrieved 23 April 2016.