DRIP-seq
DRIP-seq (DRIP-sequencing) is a technology for genome-wide profiling of a type of DNA-RNA hybrid called an "R-loop".[1] DRIP-seq utilizes a sequence-independent but structure-specific antibody for DNA-RNA immunoprecipitation (DRIP) to capture R-loops for massively parallel DNA sequencing.[1]
Introduction
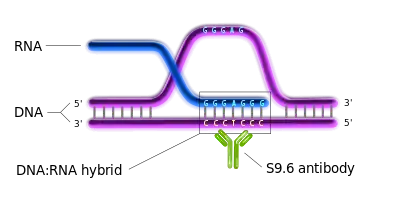
An R-loop is a three-stranded nucleic acid structure, which consists of a DNA-RNA hybrid duplex and a displaced single stranded DNA (ssDNA).[2] R-loops are predominantly formed in cytosine-rich genomic regions during transcription[2] and are known to be involved with gene expression and immunoglobulin class switching.[1][3][4] They have been found in a variety of species, ranging from bacteria to mammals.[2] They are preferentially localized at CpG island promoters in human cells and highly transcribed regions in yeast.[1][3]
Under abnormal conditions, namely elevated production of DNA-RNA hybrids, R-loops can cause genome instability by exposing single-stranded DNA to endogenous damages exerted by the action of enzymes such as AID and APOBEC, or overexposure to chemically reactive species.[4] Therefore, understanding where and in what circumstances R-loops are formed across the genome is crucial for the better understanding of genome instability. R-loop characterization was initially limited to locus specific approaches.[5] However, upon the arrival of massive parallel sequencing technologies and thereafter derivatives like DRIP-seq, the possibility to investigate entire genomes for R-loops has opened up.
DRIP-seq relies on the high specificity and affinity of the S9.6 monoclonal antibody (mAb) towards DNA-RNA hybrids of various lengths. S9.6 mAb was first created and characterized in 1986 and is currently used for the selective immunoprecipitation of R-loops.[6] Since then, it was used in diverse immunoprecipitation methods for R-loop characterization.[1][3][7][8] The concept behind DRIP-seq is similar to ChIP-sequencing; R-loop fragments are the main immunoprecipitated material in DRIP-seq.
Uses and Current Research
DRIP-seq is mainly used for genome-wide mapping of R-loops. Identifying R-loop formation sites allows the study of diverse cellular events, such as the function of R-loop formation at specific regions, the characterization of these regions, and the impact on gene expression. It can also be used to study the influence of R-loops in other processes like DNA replication and synthesis. Indirectly, DRIP-seq can be performed on mutant cell lines deficient in genes involved in R-loop resolution.[3] These types of studies provide information about the roles of the mutated gene in suppressing DNA-RNA formation and potentially about the significance of R-loops in genome instability.
DRIP-seq was first used for genome-wide profiling of R-loops in humans, which showed widespread R-loop formation at CpG island promoters.[1] Particularly, the researchers found that R-loop formation is associated with the unmethylated state of CpG islands.
DRIP-seq was later used to profile R-loop formation at transcription start and termination sites in human pluripotent Ntera2 cells.[7] In this study, the researchers revealed that R-loops on 3' ends of genes may be correlated with transcription termination.
Workflow of DRIP-seq

Genomic DNA extraction
First, genomic DNA (gDNA) is extracted from cells of interest by proteinase K treatment followed by phenol-chloroform extraction and ethanol precipitation. Additional zymolyase digestion is necessary for yeast cells to remove the cell wall prior to proteinase K treatment. gDNA can also be extracted with a variety of other methods, such as column-based methods.
Genomic DNA fragmentation
gDNA is treated with S1 nuclease to remove undesired ssDNA and RNA, followed by ethanol precipitation to remove the S1 nuclease. Then, gDNA is fragmented with restriction endonuclease, yielding double-stranded DNA (dsDNA) fragments of different sizes. Alternatively, gDNA fragments can be generated by sonication.
Immunoprecipitation
Fragmented gDNA is incubated with the DNA-RNA structure-specific S9.6 mAb. This step is unique for the DRIP-seq protocol, since it entirely relies on the high specificity and affinity of the S9.6 mAb for DNA-RNA hybrids. The antibody will recognize and bind these regions dispersed across the genome and will be used for immunoprecipitation. The S9.6 antibodies are bound to magnetic beads by interacting with specific ligands (i.e. protein A or protein G) on the surface of the beads. Thus, the DNA-RNA containing fragments will bind to the beads by means of the antibody.
Elution
The magnetic beads are washed to remove any gDNA not bound to the beads by a series of washes and DNA-RNA hybrids are recovered by elution. To remove the antibody bound to the nucleic acid hybrids, proteinase K treatment is performed followed by phenol-chloroform extraction and ethanol precipitation. This results in the isolation of purified DNA-RNA hybrids of different sizes.
Sequencing
For massive parallel sequencing of these fragments, the immunoprecipitated material is sonicated, size selected and ligated to barcoded oligonucleotide adaptors for cluster enrichment and sequencing.
Computational Analysis
To detect sites of R-loop formation, the hundreds of millions of sequencing reads from DRIP-seq are first aligned to a reference genome with a short-read sequence aligner, then peak calling methods designed for ChIP-seq can be used to evaluate DRIP signals.[1] If different cocktails of restriction enzymes were used for different DRIP-seq experiments of the same sample, consensus DRIP-seq peaks are called.[7] Typically, peaks are compared against those from a corresponding RNase H1-treated sample, which serves as an input control.[1][7]
Limitations
Due to the absence of another antibody-based method for R-loop immunoprecipitation, validation of DRIP-seq results is difficult. However, results of other R-loop profiling methods, such as DRIVE-seq, may be used to measure consensus.
On the other hand, DRIP-seq relies on existing short-read sequencing platforms for the sequencing of R-loops. In other words, all inherent limitations of these platform also apply to DRIP-seq. In particular, typical short-read sequencing platforms would produce uneven read coverage in GC-rich regions. Sequencing long R-loops might pose a challenge because R-loops are predominantly formed in cytosine-rich DNA regions. Moreover, GC-rich regions tend to have low complexity by nature, which is difficult for short read aligners to produce unique alignments.
Other R-loop Profiling Methods
Although there are several other methods for analysis and profiling of R-loop formation,[5][9][10][11][12][13][14][15][16] only few provide coverage and robustness at the genome-wide scale.[1][3]
- Non-denaturing bisulfite modification and sequencing: This method consists of bisulfite treatment followed by sequencing and relies on the mutagenic effect of sodium bisulfite on ssDNA. Although this method is primarily used to localize specific CpG island promoters,[1] it was used to detect R-loops at a minor scale[5] and other ssDNA fragile sites.[17]
- DNA:RNA In Vitro Enrichment (DRIVE-seq):[1] This method shares very similar principles of DRIP-seq except for the use of MBP-RNASEH1 endonuclease instead of the S9.6 mAb for R-loops recovery. MBP-RNASEH1 provides an alternative to S9.6 mAb when an additional capture assay is needed, however over-expression of this endonuclease may introduce cytotoxic risks in vivo.
- DNA:RNA immnunoprecipitation followed by hybridization on tiling microarray (DRIP-chip):[3] This method also relies on the use of the S9.6 mAb. However, instead of entering into a sequencing pipeline, the immunoprecipitated material in DRIP-chip is hybridized to a microarray. An advantage of DRIP-chip over DRIP-seq is the rapid obtention of the data. The limiting factors of this technique are the number of probes on the chip microarrays and absence of DNA sequence information.
References
- Ginno, PA; Lott, PL; Christensen, HC; Korf, I; Chédin, F (30 March 2012). "R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters". Molecular Cell. 45 (6): 814–25. doi:10.1016/j.molcel.2012.01.017. PMC 3319272. PMID 22387027.
- Aguilera, A; García-Muse, T (27 April 2012). "R loops: from transcription byproducts to threats to genome stability". Molecular Cell. 46 (2): 115–24. doi:10.1016/j.molcel.2012.04.009. PMID 22541554.
- Chan, YA; Aristizabal, MJ; Lu, PY; Luo, Z; Hamza, A; Kobor, MS; Stirling, PC; Hieter, P (April 2014). "Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip". PLOS Genetics. 10 (4): e1004288. doi:10.1371/journal.pgen.1004288. PMC 3990523. PMID 24743342.
- Chaudhuri, J; Tian, M; Khuong, C; Chua, K; Pinaud, E; Alt, FW (17 April 2003). "Transcription-targeted DNA deamination by the AID antibody diversification enzyme". Nature. 422 (6933): 726–30. doi:10.1038/nature01574. PMID 12692563.
- Yu, K; Chedin, F; Hsieh, CL; Wilson, TE; Lieber, MR (May 2003). "R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells". Nature Immunology. 4 (5): 442–51. doi:10.1038/ni919. PMID 12679812.
- Boguslawski, SJ; Smith, DE; Michalak, MA; Mickelson, KE; Yehle, CO; Patterson, WL; Carrico, RJ (1 May 1986). "Characterization of monoclonal antibody to DNA-RNA and its application to immunodetection of hybrids". Journal of Immunological Methods. 89 (1): 123–30. doi:10.1016/0022-1759(86)90040-2. PMID 2422282.
- Ginno, PA; Lim, YW; Lott, PL; Korf, I; Chédin, F (October 2013). "GC skew at the 5' and 3' ends of human genes links R-loop formation to epigenetic regulation and transcription termination". Genome Research. 23 (10): 1590–600. doi:10.1101/gr.158436.113. PMC 3787257. PMID 23868195.
- Skourti-Stathaki, K; Proudfoot, NJ; Gromak, N (24 June 2011). "Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination". Molecular Cell. 42 (6): 794–805. doi:10.1016/j.molcel.2011.04.026. PMC 3145960. PMID 21700224.
- El Hage, A; French, SL; Beyer, AL; Tollervey, D (15 July 2010). "Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis". Genes & Development. 24 (14): 1546–58. doi:10.1101/gad.573310. PMC 2904944. PMID 20634320.
- Duquette, ML; Huber, MD; Maizels, N (15 March 2007). "G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas". Cancer Research. 67 (6): 2586–94. doi:10.1158/0008-5472.can-06-2419. PMID 17363577.
- Huertas, P; Aguilera, A (September 2003). "Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination". Molecular Cell. 12 (3): 711–21. doi:10.1016/j.molcel.2003.08.010. PMID 14527416.
- Drolet, M; Bi, X; Liu, LF (21 January 1994). "Hypernegative supercoiling of the DNA template during transcription elongation in vitro". The Journal of Biological Chemistry. 269 (3): 2068–74. PMID 8294458.
- Gómez-González, B; Aguilera, A (15 May 2007). "Activation-induced cytidine deaminase action is strongly stimulated by mutations of the THO complex". Proceedings of the National Academy of Sciences of the United States of America. 104 (20): 8409–14. doi:10.1073/pnas.0702836104. PMC 1895963. PMID 17488823.
- Pohjoismäki, JL; Holmes, JB; Wood, SR; Yang, MY; Yasukawa, T; Reyes, A; Bailey, LJ; Cluett, TJ; Goffart, S; Willcox, S; Rigby, RE; Jackson, AP; Spelbrink, JN; Griffith, JD; Crouch, RJ; Jacobs, HT; Holt, IJ (16 April 2010). "Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid". Journal of Molecular Biology. 397 (5): 1144–55. doi:10.1016/j.jmb.2010.02.029. PMC 2857715. PMID 20184890.
- Mischo, HE; Gómez-González, B; Grzechnik, P; Rondón, AG; Wei, W; Steinmetz, L; Aguilera, A; Proudfoot, NJ (7 January 2011). "Yeast Sen1 helicase protects the genome from transcription-associated instability". Molecular Cell. 41 (1): 21–32. doi:10.1016/j.molcel.2010.12.007. PMC 3314950. PMID 21211720.
- Wahba, L; Amon, JD; Koshland, D; Vuica-Ross, M (23 December 2011). "RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability". Molecular Cell. 44 (6): 978–88. doi:10.1016/j.molcel.2011.10.017. PMC 3271842. PMID 22195970.
- Chan, K; Sterling, JF; Roberts, SA; Bhagwat, AS; Resnick, MA; Gordenin, DA (2012). "Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent". PLOS Genetics. 8 (12): e1003149. doi:10.1371/journal.pgen.1003149. PMC 3521656. PMID 23271983.