Hexaphosphabenzene
Hexaphosphabenzene is a valence isoelectronic analogue of benzene and is expected to have a similar planar structure due to resonance stabilization. Although several other allotropes of phosphorus are stable, no evidence for the existence of P6 has been reported. Preliminary ab initio calculations on the trimerisation of P2 leading to the formation of the cyclic P6 were performed, and it was predicted that hexaphosphabenzene would decompose to free P2 with an energy barrier of 13−15.4 kcal mol−1,[1] and would therefore not be observed in the uncomplexed state under normal experimental conditions. The presence of an added solvent, such as ethanol, might lead to the formation of intermolecular hydrogen bonds which may block the destabilizing interaction between phosphorus lone pairs and consequently stabilize P6.[1] The moderate barrier suggests that hexaphosphabenzene could be synthesized from a [2+2+2] cycloaddition of three P2 molecules.[2] Currently, this is a synthetic endeavour which remains to be conquered.
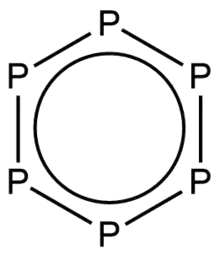 Depiction of the all-phosphorus analogue of benzene | |
| Names | |
|---|---|
| IUPAC name
hexaphosphinine | |
| Identifiers | |
3D model (JSmol) |
|
| ChemSpider | |
PubChem CID |
|
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| P6 | |
| Molar mass | 185.842571988 g·mol−1 |
| Related compounds | |
Related compounds |
Benzene, Hexazine |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
Synthesis
2(P6)).png.webp)
Isolation of hexaphosphabenzene was first achieved within a triple-decker sandwich complex in 1985 by Scherer et al. Amber coloured, air-stable crystals of [{(η5- Me5C5)Mo}2(μ,η6-P6)] are formed by reaction of [CpMo(CO)2/3]2 with excess P4 in dimethylbenzene, albeit with a yield of approximately 1%.[3][4] The crystal structure of this complex is a centrosymmetric molecule, and both five-membered rings as well as the central bridge-ligand P6 ring are planar and parallel. The average P—P distance for the hexaphosphobenzene within this complex is 2.170 Å.[3][5]
Thirty years later, Fleischmann et al. improved the synthetic yield of [{(η5- Me5C5)Mo}2(μ,η6-P6)] up to 64%. This was achieved by increasing the reaction temperature of the thermolysis of [CpMo(CO)2/3]2 with P4 to approximately 205 °C in boiling diisopropylbenzene, thus favouring the formation of [{(η5- Me5C5)Mo}2(μ,η6-P6)] as the thermodynamic product.[6]
Several analogues of this P6 triple‐decker complex where the coordinating metal and η5-ligand has been varied have also been reported. These include P6 triple‐decker complexes for Ti, V, Nb, and W, whereby the synthetic method is still based on the originally reported thermolysis of [CpM(CO)2/3]2 with P4.[7][8][9][10][11]
Electron Count

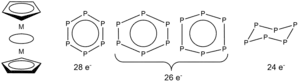
If one regards the planar P6 ring as a 6π electron donor ligand, then [{(η5- Me5C5)Mo}2(μ,η6-P6)] is a triple-decker sandwich complex with 28 valence electrons. If P6, similar to C6H6, is taken as a 10π electron donor, a 32 valence electron count may be obtained. In most triple-decker complexes with an electron count ranging from 26 to 34, the structure of the middle ring is planar ([{(η5- Cp)M}2(μ,η6-P6)] with M = Mo, Sc, Y, Zr, Hf, V, Nb, Ta, Cr, and W).[12][13] In the 24 valence electron [{(η5- Cp)Ti}2(μ,η6-P6)] complex, however, a distortion is observed, and the P6 ring is puckered.[7]
Calculations have concluded that completely filled 2a*and 2b* orbitals in 28 valence electron complexes lead to a planar symmetrical P6 middle ring. In 26 valence electron complexes, the occupancy of either 2a*or 2b* results in in-plane or bisallylic distortions and an asymmetric planar middle ring. The puckering of P6 in 24 valence electron complexes is due to the stabilization of 5a, as well as that conferred by the tetravalent oxidation state of Ti in [{(η5- Cp)Ti}2(μ,η6-P6)].[7][14]
Reactivity
Mo)2(%CE%BC%252C%CE%B76-P6).png.webp)
One-electron oxidation
The reactivity of [{(η5- Me5C5)Mo}2(μ,η6-P6)] toward silver and copper monocationic salts of the weakly coordinating anion [Al{OC(CF3)3}4]− ([TEF]) was studied by Fleischmann et al. in 2015.[6] Addition of a solution of Ag[TEF] or Cu[TEF] to a solution of [{(η5- Me5C5)Mo}2(μ,η6-P6)] in chloroform results in oxidation of the complex, which can be observed by an immediate colour change from amber to dark teal. The magnetic moment of the dark teal crystals determined by the Evans NMR method is equal to 1.67 μB, which is consistent with one unpaired electron. Accordingly, [{(η5- Me5C5)Mo}2(μ,η6-P6)]+ is detected by ESI mass spectrometry.
The crystal structure of the teal product shows that the triple‐decker geometry is retained during the one‐electron oxidation of [{(η5- Me5C5)Mo}2(μ,η6-P6)]. The Mo—Mo bond length of the [{(η5- Me5C5)Mo}2(μ,η6-P6)]+ cation is 2.6617(4) Å; almost identical to the bond length determined for the unoxidized species at 2.6463(3) Å. However, the P—P bond lengths are strongly affected by the oxidation. While the P1—P1′ and P3—P3′ bonds are elongated, the remaining P—P bonds are shortened compared to the average P—P bond length of about 2.183 Å in the unoxidized species. Therefore, the middle deck of the 27 valence electron [{(η5- Me5C5)Mo}2(μ,η6-P6)]+ complex can best be described as a bisallylic distorted P6 ligand, intermediate between the 28 valence electron complexes with a perfectly planar symmetrical ring, and those with 26 valence electrons displaying a more amplified in-plane distortion. Density functional theorem (DFT) calculations confirm that this distortion is due to depopulation of the P bonding orbitals upon oxidation of the triple-decker sandwich complex.[6]
Cu[TEF] & Ag[TEF]
Mo)2(%CE%BC%252C%CE%B76-P6)).png.webp)
To avoid oxidation of [{(η5- Me5C5)Mo}2(μ,η6-P6)], further reactions were performed in toluene to decrease the redox potential of the cations. This resulted in a bright orange coordination product upon reaction with copper, although a mixture also containing the dark teal oxidation product was obtained upon reaction with silver.
Single‐crystal X‐ray analysis reveals that this product displays a distorted square‐planar coordination environment around the central cation through two side‐on coordinating P—P bonds. The Ag—P distances are approximately 2.6 Å, whereas the Cu—P distances are determined to be approximately 2.4 Å. The P—P bonds are therefore elongated to 2.2694(16) Å and 2.2915(14) Å upon coordination to copper and silver, respectively, whilst the remaining P—P bonds are unaffected.
In another experiment Cu[TEF] is treated with [{(η5- Me5C5)Mo}2(μ,η6-P6)] in pure toluene and the solution shows the bright orange color of the complex cation [Cu([{(η5- Me5C5)Mo}2(μ,η6-P6)])2]+. However, analysis of crystals from this solution reveals a distorted tetrahedral coordination environment around Cu. The resulting Cu—P distances are somewhat shorter than their counterparts discussed above. The coordinating P—P bonds are a little longer, which is attributed to less steric crowding in the tetrahedral coordination geometry around the Cu center.
The successful isolation of [Cu([{(η5- Me5C5)Mo}2(μ,η6-P6)])2]+ either as its tetrahedral or square‐planar isomer is therefore achievable. DFT calculations show that the enthalpy for the tetrahedral to square‐planar isomerization is positive for both metals, with the tetrahedral coordination being favored. When entropy is taken into account, small positive values for Cu+ and larger, but negative, values for Ag+ are observed. This means that the tetrahedral geometry is predominant for Cu+, but a significant percentage of the complexes adopt a square‐planar geometry in solution. For Ag+, the equilibrium is shifted significantly to the right side, which is presumably why a tetrahedral coordination of [{(η5- Me5C5)Mo}2(μ,η6-P6)] and Ag+ has not yet been observed.
Examination of the crystal packing reveals that these products are layered compounds that crystallize in the monoclinic C2/c space group with alternating negatively charged layers of the [TEF] anions and positively charged layers of isolated [M([{(η5- Me5C5)Mo}2(μ,η6-P6)])2]+ complexes. The layers lie inside the bc plane, alternate along the a axis, and do not form a two‐dimensional network.[6]
Tl[TEF]
The treatment of [{(η5- Me5C5)Mo}2(μ,η6-P6)] with Tl[TEF] in chloroform gives an immediate color change from amber to a deep red. The crystal structure reveals a trigonal pyramidal coordination of the thallium cation, Tl+, by three side‐on coordinating P—P bonds of the P6 ligands. Two of these P6 ligands show shorter and uniform Tl—P distances of 3.2–3.3 Å with P—P bonds elongated to about 2.22 Å, whilst the third unit shows an unsymmetrical coordination with long Tl—P distances of approximately 3.42 and 3.69 Å and no P—P bond elongation.
Mo)2(%CE%BC%252C%CE%B76-P6)))2)%252B_and_(Tl((((%CE%B75-_Me5C5)Mo)2(%CE%BC%252C%CE%B76-P6)))2)%252B.png.webp)
Although the environment of Tl+ is distinctly different from that of Cu+ and Ag+, their structures are related by the two‐dimensional coordination network that propagates inside the bc plane. Crucially, whilst Cu+ and Ag+ form layered structures with isolated [M([{(η5- Me5C5)Mo}2(μ,η6-P6)])2]+ complex cations, there is a statistical distribution of the Tl+ cations inside the two‐dimensional coordination, which shows further interconnection of the P6 ligands to form an extended 2D network that could be regarded as a supramolecular analogue of graphene.[6]
Jahn-Teller Distortion
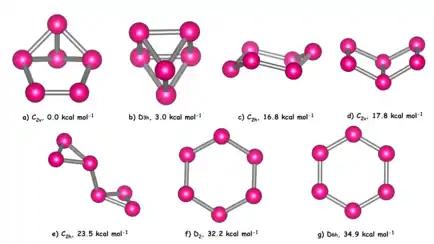
Despite the triple-decker sandwich complex {(η5-Me5C5)Mo}2(μ,η6-P6) containing a demonstrably planar P6 ring with equal P—P bond lengths, theoretical calculations reveal that there are at least 7 non-planar P6 isomers lower in energy than the planar benzene-like D6h structure.[1][2][15][16][17][18][19][20][21][22][23][24] In increasing order of energy these are: benzvalene, prismane, chair, Dewar benzene, bicyclopropenyl, distorted benzene, and benzene.[24]
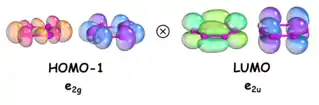
A pseudo Jahn-Teller effect (PJT) is responsible for distortion of the D6h benzene-like structure into the D2 structure,[25][26][27][28][29][30] which occurs along the e2u doubly degenerate mode as a result of vibronic coupling of the HOMO − 1 (e2g) and LUMO (e2u): e2g ⊗ e2u = a1u ⊕ a2u ⊕ e2u. The distorted structure is calculated to lie just 2.7 kcal mol−1 lower in energy than the D6h structure. If the uncomplexed structure were to be successfully synthesized, the aromaticity of the benzene-like P6 structure would not be sufficient to stabilize the planar geometry, and the PJT effect would result in distortion of the ring.[31]
Isomers
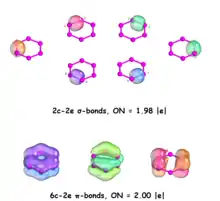
Adaptive Natural Density Partitioning (AdNDP) is a theoretical tool developed by Alexander Boldyrev that is based on the concept of the electron pair as the main element of chemical bonding models. It can therefore recover Lewis bonding elements such as 1c–2e core electrons and lone pairs, 2c–2e objects which are two-center two-electron bonds, as well as delocalized many-center bonding elements with respect to aromaticity.
The AdNDP analysis of the seven representative low-lying P6 structures structures reveal that these are well described by the classical Lewis model. A lone pair on each phosphorus atom, a two-center-two-electron (2c–2e) σ-bond in every pair of adjacent P atoms, and an additional 2c–2e π-bond between adjacent 2-coordinated P atoms are found, with occupation numbers (ON) of all these bonding elements above 1.92 |e|.[31]
The chemical bonding in the chair structure is unusual. Based on fragment orbital analysis, it was concluded that two linkages between the two P3 fragments are of the one-electron hemibond type. The AdNDP analysis reveals a lone pair on each P atom and six 2c–2e P—P σ-bonds. One 3c–2e π-bond in every P3 triangle was revealed with the user-directed form of the AdNDP analysis, as well as a 4c–2e bond responsible for bonding between the two P3 triangle, confirming that this isomer cannot be represented by a single Lewis structure, and requires a resonance of two Lewis structures, or can be described by a single formula with delocalized bonding elements.
Both the D6h benzene-like structure, as well as the D2 isomer of P6 is similar to the reported AdNDP bonding pattern of the C6H6 benzene molecule:[32] 2c–2e σ-bond and lone pairs, as well as delocalized 6c-2e π-bonds. The distortion due to the PJT effect therefore does not significantly disturb the bonding picture.[31]
Suppression
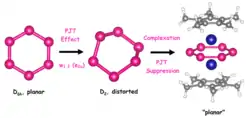

The planar P6 hexagonal structure D6h is a second-order saddle point due to the pseudo-Jahn-Teller effect (PJT), which leads to the D2 distorted structure. Upon sandwich complex formation the PJT effect is suppressed due to filling of the unoccupied molecular orbitals involved in vibronic coupling in P6 with electron pairs of Mo atoms.[33][34][35] Specifically, from molecular orbital analysis it was determined that, upon complex formation, the LUMO in the isolated P6 structure is now occupied in the triple-decker complex as a result of the appreciable δ-type M → L back-donation mechanism from the occupied dx2–y2 and dxy atomic orbitals of the Mo atom into the partially antibonding π molecular orbitals of P6, thus restoring the high symmetry and planarity of P6.[35]
References
- Nguyen, Minh Tho; Hegarty, Anthony F. (1986-01-01). "Can the cyclic hexaphosphabenzene (P6) exist?". Journal of the Chemical Society, Chemical Communications (5): 383–385. doi:10.1039/C39860000383. ISSN 0022-4936.
- Nagase, Shigeru; Ito, Keiji (1986-04-25). "Theoretical study of hexaphosphabenzene and its valence isomers. Is cyclic P6 stable?". Chemical Physics Letters. 126 (1): 43–47. Bibcode:1986CPL...126...43N. doi:10.1016/0009-2614(86)85113-2. ISSN 0009-2614.
- Scherer, Otto J.; Sitzmann, Helmut; Wolmershäuser, Gotthelf (1985). "Hexaphosphabenzene as Complex Ligand". Angewandte Chemie International Edition in English. 24 (4): 351–353. doi:10.1002/anie.198503511. ISSN 1521-3773.
- Meier, Herbert; Zeller, Klaus-Peter (1975). "The Wolff Rearrangement of α-Diazo Carbonyl Compounds". Angewandte Chemie International Edition in English. 14 (1): 32–43. doi:10.1002/anie.197500321. ISSN 1521-3773.
- Scherer, Otto J. (2000). "Small Neutral Pn Molecules". Angewandte Chemie International Edition. 39 (6): 1029–1030. doi:10.1002/(SICI)1521-3773(20000317)39:6<1029::AID-ANIE1029>3.0.CO;2-6. ISSN 1521-3773. PMID 10760912.
- Fleischmann, Martin; Dielmann, Fabian; Gregoriades, Laurence J.; Peresypkina, Eugenia V.; Virovets, Alexander V.; Huber, Sebastian; Timoshkin, Alexey Y.; Balázs, Gábor; Scheer, Manfred (2015). "Redox and Coordination Behavior of the Hexaphosphabenzene Ligand in [(Cp*Mo)2(μ,η6:η6-P6)] Towards the "Naked" Cations Cu+, Ag+, and Tl+". Angewandte Chemie International Edition. 54 (44): 13110–13115. doi:10.1002/anie.201506362. ISSN 1521-3773. PMC 4675074. PMID 26337857.
- Scherer, Otto J.; Swarowsky, Herbert; Wolmershäuser, Gotthelf; Kaim, Wolfgang; Kohlmann, Stephan (1987). "[(η5-C5Me5)2 Ti2P6], a Distorted Dimetallaphosphacubane". Angewandte Chemie International Edition in English. 26 (11): 1153–1155. doi:10.1002/anie.198711531. ISSN 1521-3773.
- Herberhold, Max; Frohmader, Gudrun; Milius, Wolfgang (1996-09-20). "Neue vanadium-komplexe mit substituentenfreien phosphorliganden". Journal of Organometallic Chemistry (in German). 522 (2): 185–196. doi:10.1016/0022-328X(96)06305-X. ISSN 0022-328X.
- Scherer, Otto J.; Schwalb, Joachim; Swarowsky, Herbert; Wolmershäuser, Gotthelf; Kaim, Wolfgang; Gross, Renate (1988-03-01). "Tripeldecker-Sandwichkomplexe mit cyclo-P6-Mitteldeck". Chemische Berichte. 121 (3): 443–449. doi:10.1002/cber.19881210309. ISSN 0009-2940.
- Scherer, Otto J.; Vondung, Jürgen; Wolmershäuser, Gotthelf (1989). "Tetraphosphacyclobutadiene as Complex Ligand". Angewandte Chemie International Edition in English. 28 (10): 1355–1357. doi:10.1002/anie.198913551. ISSN 1521-3773.
- Reddy, A. Chandrasekhar; Jemmis, Eluvathingal D.; Scherer, Otto J.; Winter, Rainer; Heckmann, Gert; Wolmershaeuser, Gotthelf (1992-11-01). "Electronic structure of triple-decker sandwich complexes with P6 middle rings. Synthesis and x-ray structure determination of bis(.eta.5-1,3-di-tert-butylcyclopentadienyl)(.mu.-.eta.6:.eta.6-hexaphosphorin)diniobium". Organometallics. 11 (11): 3894–3900. doi:10.1021/om00059a064. ISSN 0276-7333.
- Jemmis, Eluvathingal D.; Reddy, A. Chandrasekhar. (1988-07-01). "Electronic structure of triple-decker sandwich compounds with P5, P6, As5, and CnHn as middle rings". Organometallics. 7 (7): 1561–1564. doi:10.1021/om00097a019. ISSN 0276-7333.
- Jemmis, Eluvathingal D.; Reddy, A. Chandrasekhar (1990-06-01). "Structure and bonding in metallocenes and oligomers" (PDF). Proceedings of the Indian Academy of Sciences - Chemical Sciences. 102 (3): 379–393. doi:10.1007/BF02841950 (inactive 2021-01-19). ISSN 0973-7103.CS1 maint: DOI inactive as of January 2021 (link)
- Rani, Dandamudi Usha; Prasad, Dasari L. V. K.; Nixon, John F.; Jemmis, Eluvathingal D. (2007). "Electronic structure and bonding studies on triple-decker sandwich complexes with a P6 middle ring". Journal of Computational Chemistry. 28 (1): 310–319. doi:10.1002/jcc.20521. ISSN 1096-987X. PMID 17103397. S2CID 25386398.
- Jones, R.; Hohl, D. (1990). "Structure of phosphorus clusters using simulated annealing—P2 to P8". The Journal of Chemical Physics. 92 (11): 6710–6721. Bibcode:1990JChPh..92.6710J. doi:10.1063/1.458306. S2CID 31227289.
- Jones, R. O.; Ganteför, G.; Hunsicker, S.; Pieperhoff, P. (1995-12-08). "Structure and spectroscopy of phosphorus cluster anions: Theory (simulated annealing) and experiment (photoelectron detachment)". The Journal of Chemical Physics. 103 (22): 9549–9562. Bibcode:1995JChPh.103.9549J. doi:10.1063/1.469969. ISSN 0021-9606.
- Warren, D. Scott; Gimarc, Benjamin M. (1992-06-01). "Valence isomers of benzene and their relationship to isomers of isoelectronic P6". Journal of the American Chemical Society. 114 (13): 5378–5385. doi:10.1021/ja00039a058. ISSN 0002-7863.
- Olvera de la Cruz, M.; Mayes, A. M.; Swift, B. W. (1992-01-01). "Transition to lamellar-catenoid structure in block-copolymer melts". Macromolecules. 25 (2): 944–948. Bibcode:1992MaMol..25..944O. doi:10.1021/ma00028a067. ISSN 0024-9297.
- Haeser, Marco; Schneider, Uwe; Ahlrichs, Reinhart (1992-11-01). "Clusters of phosphorus: a theoretical investigation". Journal of the American Chemical Society. 114 (24): 9551–9559. doi:10.1021/ja00050a039. ISSN 0002-7863.
- Jones, J. L.; Olvera de la Cruz, M. (1994-04-01). "Transitions to periodic structures: Higher harmonic corrections with concentration fluctuations". The Journal of Chemical Physics. 100 (7): 5272–5279. Bibcode:1994JChPh.100.5272J. doi:10.1063/1.467191. ISSN 0021-9606.
- Häser, Marco; Treutler, Oliver (1995-03-01). "Calculated properties of P2, P4, and of closed‐shell clusters up to P18". The Journal of Chemical Physics. 102 (9): 3703–3711. Bibcode:1995JChPh.102.3703H. doi:10.1063/1.468552. ISSN 0021-9606.
- Kobayashi, Kaoru; Miura, Hideki; Nagase, Shigeru (1994-07-20). "The heavier group 15 analogues of benzene, cyclobutadiene and their valence isomers (M6 and M4, M = P, As, Sb and Bi)". Journal of Molecular Structure: THEOCHEM. 311: 69–77. doi:10.1016/S0166-1280(09)80043-2. ISSN 0166-1280.
- Ballone, P.; Jones, R. O. (1994-04-01). "Density functional study of phosphorus and arsenic clusters using local and nonlocal energy functionals". The Journal of Chemical Physics. 100 (7): 4941–4946. Bibcode:1994JChPh.100.4941B. doi:10.1063/1.467213. ISSN 0021-9606.
- Hiberty, Philippe C.; Volatron, François (2007). "Ab initio conformational study of the P6 potential surface: Evidence for a low-lying one-electron-bonded isomer". Heteroatom Chemistry. 18 (2): 129–134. doi:10.1002/hc.20324. ISSN 1098-1071.
- Bersuker, Isaac B. (2001-04-01). "Modern Aspects of the Jahn−Teller Effect Theory and Applications To Molecular Problems". Chemical Reviews. 101 (4): 1067–1114. doi:10.1021/cr0004411. ISSN 0009-2665. PMID 11709858.
- Bersuker, I. B. The Jahn-Teller effect. Cambridge. ISBN 978-0-511-52476-9. OCLC 967605700.
- Isaak Borisovich Bersuker (2008). The Jahn-Teller Effect and Beyond: Selected Works of Isaac Bersuker with Commentaries : Dedicated to Isaac B. Bersuker on Occasion of His 80th Birthday. Academy of Sciences of Moldova. ISBN 978-9975-62-212-7.
- Pearson, Ralph G. (1975-06-01). "Concerning Jahn-Teller Effects". Proceedings of the National Academy of Sciences. 72 (6): 2104–2106. Bibcode:1975PNAS...72.2104P. doi:10.1073/pnas.72.6.2104. ISSN 0027-8424. PMC 432704. PMID 16592247.
- Bersuker, I. B. (1966-04-01). "On the origin of ferroelectricity in perovskite-type crystals". Physics Letters. 20 (6): 589–590. Bibcode:1966PhL....20..589B. doi:10.1016/0031-9163(66)91127-9. ISSN 0031-9163.
- Öpik, U.; Pryce, Maurice Henry Lecorney (1957-01-29). "Studies of the Jahn-Teller effect. I. A survey of the static problem". Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences. 238 (1215): 425–447. Bibcode:1957RSPSA.238..425O. doi:10.1098/rspa.1957.0010. S2CID 119862339.
- Galeev, Timur R.; Boldyrev, Alexander I. (2011-11-15). "Planarity takes over in the CxHxP6−x (x = 0–6) series at x = 4". Physical Chemistry Chemical Physics. 13 (46): 20549–20556. Bibcode:2011PCCP...1320549G. doi:10.1039/C1CP21959F. ISSN 1463-9084. PMID 21869975.
- Zubarev, Dmitry Yu.; Boldyrev, Alexander I. (2008-12-05). "Revealing Intuitively Assessable Chemical Bonding Patterns in Organic Aromatic Molecules via Adaptive Natural Density Partitioning". The Journal of Organic Chemistry. 73 (23): 9251–9258. doi:10.1021/jo801407e. ISSN 0022-3263. PMID 18980326.
- Sergeeva, Alina P.; Boldyrev, Alexander I. (2010-09-13). "Flattening a Puckered Pentasilacyclopentadienide Ring by Suppression of the Pseudo Jahn−Teller Effect". Organometallics. 29 (17): 3951–3954. doi:10.1021/om1006038. ISSN 0276-7333.
- Pokhodnya, Konstantin; Olson, Christopher; Dai, Xuliang; Schulz, Douglas L.; Boudjouk, Philip; Sergeeva, Alina P.; Boldyrev, Alexander I. (2011-01-07). "Flattening a puckered cyclohexasilane ring by suppression of the pseudo-Jahn–Teller effect". The Journal of Chemical Physics. 134 (1): 014105. Bibcode:2011JChPh.134a4105P. doi:10.1063/1.3516179. ISSN 0021-9606. PMID 21218995.
- Ivanov, Alexander S.; Bozhenko, Konstantin V.; Boldyrev, Alexander I. (2012-08-20). "On the Suppression Mechanism of the Pseudo-Jahn–Teller Effect in Middle E6 (E = P, As, Sb) Rings of Triple-Decker Sandwich Complexes". Inorganic Chemistry. 51 (16): 8868–8872. doi:10.1021/ic300786w. ISSN 0020-1669. PMID 22845625.