Immunolabeling
Immunolabeling is a biochemical process that enables the detection and localization of an antigen to a particular site within a cell, tissue, or organ. Antigens are organic molecules, usually proteins, capable of binding to an antibody. These antigens can be visualized using a combination of antigen-specific antibody as well as a means of detection, called a tag, that is covalently linked to the antibody.[1] If the immunolabeling process is meant to reveal information about a cell or its substructures, the process is called immunocytochemistry.[2] Immunolabeling of larger structures is called immunohistochemistry.[3]
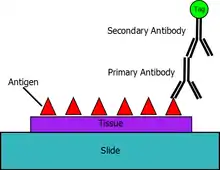
There are two complex steps in the manufacture of antibody for immunolabeling. The first is producing the antibody that binds specifically to the antigen of interest and the second is fusing the tag to the antibody. Since it is impractical to fuse a tag to every conceivable antigen-specific antibody, most immunolabeling processes use an indirect method of detection. This indirect method employs a primary antibody that is antigen-specific and a secondary antibody fused to a tag that specifically binds the primary antibody. This indirect approach permits mass production of secondary antibody that can be bought off the shelf.[4] Pursuant to this indirect method, the primary antibody is added to the test system. The primary antibody seeks out and binds to the target antigen. The tagged secondary antibody, designed to attach exclusively to the primary antibody, is subsequently added.
Typical tags include: a fluorescent compound, gold beads, a particular epitope tag,[5] or an enzyme that produces a colored compound. The association of the tags to the target via the antibodies provides for the identification and visualization of the antigen of interest in its native location in the tissue, such as the cell membrane, cytoplasm, or nuclear membrane. Under certain conditions the method can be adapted to provide quantitative information.[4]
Immunolabeling can be used in pharmacology, molecular biology, biochemistry and any other field where it is important to know of the precise location of an antibody-bindable molecule.[6][7][8]
Indirect vs. direct method
There are two methods involved in immunolabeling, the direct and the indirect methods. In the direct method of immunolabeling, the primary antibody is conjugated directly to the tag.[9] The direct method is useful in minimizing cross-reaction, a measure of nonspecificity that is inherent in all antibodies and that is multiplied with each additional antibody used to detect an antigen. However, the direct method is far less practical than the indirect method, and is not commonly used in laboratories, since the primary antibodies must be covalently labeled, which require an abundant supply of purified antibody. Also, the direct method is potentially far less sensitive than the indirect method.[10] Since several secondary antibodies are capable of binding to different parts, or domains, of a single primary antibody binding the target antigen, there is more tagged antibody associated with each antigen. More tag per antigen results in more signal per antigen.[11]
Different indirect methods can be employed to achieve high degrees of specificity and sensitivity. First, two-step protocols are often used to avoid the cross-reaction between the immunolabeling of multiple primary and secondary antibody mixtures, where secondary fragment antigen-binding antibodies are frequently used. Secondly, haptenylated primary antibodies can be used, where the secondary antibody can recognize the associated hapten. The hapten is covalently linked to the primary antibody by succinyl imidesters or conjugated IgG Fc-specific Fab sections. Lastly, primary monoclonal antibodies that have different Ig isotypes can be detected by specific secondary antibodies that are against the isotype of interest.[10]
Antibody binding and specificity
Overall, antibodies must bind to the antigens with a high specificity and affinity.[12] The specificity of the binding refers to an antibody's capacity to bind and only bind a single target antigen. Scientists commonly use monoclonal antibodies and polyclonal antibodies, which are composed of synthetic peptides. During the manufacture of these antibodies, antigen specific antibodies are sequestered by attaching the antigenic peptide to an affinity column and allowing nonspecific antibody to simply pass through the column. This decreases the likelihood that the antibodies will bind to an unwanted epitope of the antigen not found on the initial peptide. Hence, the specificity of the antibody is established by the specific reaction with the protein or peptide that is used for immunization by specific methods, such as immunoblotting or immunoprecipitation.[13]
In establishing the specificity of antibodies, the key factor is the type of synthetic peptides or purified proteins being used. The lesser the specificity of the antibody, the greater the chance of visualizing something other than the target antigen. In the case of synthetic peptides, the advantage is the amino acid sequence is easily accessible, but the peptides do not always resemble the 3-D structure or post-translational modification found in the native form of the protein. Therefore, antibodies that are produced to work against a synthetic peptide may have problems with the native 3-D protein. These types of antibodies would lead to poor results in immunoprecipitation or immunohistochemistry experiments, yet the antibodies may be capable of binding to the denatured form of the protein during an immunoblotting run. On the contrary, if the antibody works well for purified proteins in their native form and not denatured, an immunoblot cannot be used as a standardized test to determine the specificity of the antibody binding, particularly in immunohistochemistry.[14]
Specific immunolabeling techniques
Immunolabeling for light microscopy
Light microscopy is the use of a light microscope, which is an instrument that requires the usage of light to view the enlarged specimen. In general, a compound light microscope is frequently used, where two lenses, the eyepiece, and the objective work simultaneously to generate the magnification of the specimen.[15] Light microscopy frequently uses immunolabeling to observe targeted tissues or cells. For instance, a study was conducted to view the morphology and the production of hormones in pituitary adenoma cell cultures via light microscopy and other electron microscopic methods. This type of microscopy confirmed that the primary adenoma cell cultures keep their physiological characteristics in vitro, which matched the histology inspection. Moreover, cell cultures of human pituitary adenomas were viewed by light microscopy and immunocytochemistry, where these cells were fixed and immunolabeled with a monoclonal mouse antibody against human GH and a polyclonal rabbit antibody against PRL. This is an example of how a immunolabeled cell culture of pituitary adenoma cells that were viewed via light microscopy and by other electron microscopy techniques can assist with the proper diagnosis of tumors.[16]
Immunolabeling for electron microscopy
Electron microscopy (EM) is a focused area of science that uses the electron microscope as a tool for viewing tissues.[17] Electron microscopy has a magnification level up to 2 million times, whereas light microscopy only has a magnification up to 1000-2000 times.[18] There are two types of electron microscopes, the transmission electron microscope and the scanning electron microscope.[17]
Electron microscopy is a common method that uses the immunolabeling technique to view tagged tissues or cells. The electron microscope method follows many of the same concepts as immunolabeling for light microscopy, where the particular antibody is able to recognize the location of the antigen of interest and then be viewed by the electron microscope. The advantage of electron microscopy over light microscopy is the ability to view the targeted areas at their subcellular level. Generally, a heavy metal that is electron dense is used for EM, which can reflect the incident electrons. Immunolabeling is typically confirmed using the light microscope to assure the presence of the antigen and then followed up with the electron microscope.[19]
Immunolabeling and electron microscopy are often used to view chromosomes. A study was conducted to view possible improvements of immunolabeling chromosome structures, such as topoisomerase IIα and condensin in dissected mitotic chromosomes. In particular, these investigators used UV irradiation of separated nuclei or showed how chromosomes assist by high levels of specific immunolabeling, which were viewed by electron microscopy.[20]
Immunolabeling for transmission electron microscopy
Transmission electron microscopy (TEM) uses a transmission electron microscope to form a two-dimensional image by shooting electrons through a thin piece of tissue. The brighter certain areas are on the image, the more electrons that are able to move through the specimen.[17] Transmission Electron Microscopy has been used as a way to view immunolabeled tissues and cells. For instance, bacteria can be viewed by TEM when immunolabeling is applied. A study was conducted to examine the structures of CS3 and CS6 fimbriae in different Escherichia coli strains, which were detected by TEM followed by negative staining, and immunolabeling. More specifically, immunolabeling of the fimbriae confirmed the existence of different surface antigens.[21]
Immunolabeling for scanning electron microscopy
Scanning electron microscopy (SEM) uses a scanning electron microscope, which produces large images that are perceived as three-dimensional when, in fact, they are not. This type of microscope concentrates a beam of electrons across a very small area (2-3 nm) of the specimen in order to produce electrons from said specimen. These secondary electrons are detected by a sensor, and the image of the specimen is generated over a certain time period.[17]
Scanning electron microscopy is a frequently used immunolabeling technique. SEM is able to detect the surface of cellular components in high resolution. This immunolabeling technique is very similar to the immuno-fluorescence method, but a colloidal gold tag is used instead of a fluorophore. Overall, the concepts are very parallel in that an unconjugated primary antibody is used and sequentially followed by a tagged secondary antibody that works against the primary antibody.[22] Sometimes SEM in conjunction with gold particle immunolabeling is troublesome in regards to the particles and charges resolution under the electron beam; however, this resolution setback has been resolved by the improvement of the SEM instrumentation by backscattered electron imaging.[23] This is because electron backscattered diffraction patterns provide a clean surface of the sample to interact with the primary electron beam.[24]
Immunolabeling with gold (Immunogold Labeling)
Immunolabeling with gold particles, also known as immunogold staining, is used regularly with scanning electron microscopy and transmission electron microscopy to successfully identify the area within cells and tissues where antigens are located.[23] The gold particle labeling technique was first published by Faulk, W. and Taylor, G. when they were able to tag gold particles to anti-salmonella rabbit gamma globulins in one step in order to identify the location of the antigens of salmonella.[23][25]
Studies have shown that the size of the gold particle must be enlarged (>40 nm) to view the cells in low magnification, but gold particles that are too large can decrease the efficiency of the binding of the gold tag. Scientists have concluded the usage of smaller gold particles (1-5 nm) should be enlarged and enhanced with silver. Although osmium tetroxide staining can scratch the silver, gold particle enhancement was found not to be susceptible to scratching by osmium tetroxide staining; therefore, many cell adhesion studies of different substrates can use the immunogold labeling mechanism via the enhancement of the gold particles.[26]
References
- Hyatt, A.D., & Wise, T.G. (2001). "Immunolabeling". Immunocytochemistry & In Situ Hybridization in the Biomedical Sciences - Chapter 5 - Immunolabeling. Boston, MA: Birkhauser Boston. pp. 73–107. doi:10.1007/978-1-4612-0139-7_5. ISBN 978-1-46-12-0139-7.
- Nanci A, Wazen R, Nishio C, Zalzal SF (2008). "Immunocytochemistry of matrix proteins in calcified tissues: functional biochemistry on section". Eur J Histochem. 52 (4): 201–14. doi:10.4081/1218. PMID 19109094.
- Swanson PE (September 1988). "Foundations of immunohistochemistry. A practical review". Am. J. Clin. Pathol. 90 (3): 333–9. doi:10.1093/ajcp/90.3.333. PMID 3046324.
- Rapley R, Walker JM, eds. (2008). Molecular biomethods handbook (2nd ed.). Totowa, NJ: Humana Press. p. 1066. ISBN 978-1-60327-370-1.
- Pang J, Zeng X, Xiao RP, Lakatta EG, Lin L (June 2009). "Design, generation, and testing of mammalian expression modules that tag membrane proteins". Protein Sci. 18 (6): 1261–71. doi:10.1002/pro.136. PMC 2774436. PMID 19472344.
- Rangell LK, Keller GA (August 2000). "Application of microwave technology to the processing and immunolabeling of plastic-embedded and cryosections". Journal of Histochemistry and Cytochemistry. 48 (8): 1153–9. doi:10.1177/002215540004800812. PMID 10898808.
- Malecki M, Hsu A, Truong L, Sanchez S (January 2002). "Molecular immunolabeling with recombinant single-chain variable fragment (scFv) antibodies designed with metal-binding domains". Proceedings of the National Academy of Sciences of the United States of America. 99 (1): 213–8. Bibcode:2002PNAS...99..213M. doi:10.1073/pnas.261567298. PMC 117541. PMID 11756693.
- Haycock JW (September 1989). "Quantitation of tyrosine hydroxylase, protein levels: spot immunolabeling with an affinity-purified antibody". Analytical Biochemistry. 181 (2): 259–66. doi:10.1016/0003-2697(89)90240-6. PMID 2573292.
- Chandler, Douglas E.; Roberson, Robert W. (2009). Bioimaging : current concepts in light and electron microscopy. Sudbury, Mass.: Jones and Bartlett Publishers. p. 311. ISBN 978-0-7637-3874-7.
- Buchwalow IB, Minin EA, Boecker W (2005). "A multicolor fluorescence immunostaining technique for simultaneous antigen targeting". Acta Histochem. 107 (2): 143–8. doi:10.1016/j.acthis.2005.01.003. PMID 15950054.
- Ramos-Vara JA (July 2005). "Technical aspects of immunohistochemistry". Vet. Pathol. 42 (4): 405–26. doi:10.1354/vp.42-4-405. PMID 16006601. S2CID 6229029.
- Kumagai I, Tsumoto K (2010-04-19). Antigen-Antibody Binding. eLS. pp. 1–7. doi:10.1002/9780470015902.a0001117.pub2. ISBN 978-0470016176.
- Burry RW (February 2000). "Specificity controls for immunocytochemical methods". J. Histochem. Cytochem. 48 (2): 163–6. doi:10.1177/002215540004800201. PMID 10639482.
- Bordeaux J, Welsh A, Agarwal S, Killiam E, Baquero M, Hanna J, Anagnostou V, Rimm D (March 2010). "Antibody validation". BioTechniques. 48 (3): 197–209. doi:10.2144/000113382. PMC 3891910. PMID 20359301.
- Murphy, Douglas B. Davidson; Michael W. (2013). Fundamentals of Light Microscopy and Electronic Imaging (Second ed.). Hoboken, NJ: John Wiley and Sons, Inc. pp. 1–2. ISBN 978-0-471-69214-0.
- Fazekas I, Hegedüs B, Bácsy E, et al. (2005). "Characterization of human pituitary adenomas in cell cultures by light and electron microscopic morphology and immunolabeling". Folia Histochemica et Cytobiologica. 43 (2): 81–90. PMID 16044945.
- Russell, John J. Bozzola; Lonnie D. (1999). Electron Microscopy: Principles and Techniques for Biologists (2nd Edition) (2. ed.). Sudbury, Mass. [u.a.]: Jones and Bartlett. pp. 5 & 9. ISBN 978-0-7637-0192-5.
- Kim, Oliver (2009-01-22). "Electron Microscopes vs. Optical (Light) Microscopes". Microbehunter Microscopy Magazine. Retrieved 2013-05-04.
- George, Andrew J.T.; Urch, Catherine E. (2000). Diagnostic and Therapeutic Antibodies. Totowa, N.J.: Humana Press. pp. 439–452. ISBN 9781592590766.
- Maeshima K, Eltsov M, Laemmli UK (November 2005). "Chromosome structure: improved immunolabeling for electron microscopy" (PDF). Chromosoma. 114 (5): 365–75. doi:10.1007/s00412-005-0023-7. PMID 16175370. S2CID 14768368.
- Lüdi S, Frey J, Favre D, Stoffel MH (April 2006). "Assessing the expression of enterotoxigenic Escherichia coli-specific surface antigens in recombinant strains by transmission electron microscopy and immunolabeling". J. Histochem. Cytochem. 54 (4): 473–7. doi:10.1369/jhc.5A6794.2005. PMID 16344328.
- Goldberg MW (2008). Immunolabeling for scanning electron microscopy (SEM) and field emission SEM. Methods Cell Biol. Methods in Cell Biology. 88. pp. 109–30. doi:10.1016/S0091-679X(08)00407-X. ISBN 9780123743206. PMID 18617031.
- Rosso F, Papale F, Barbarisi A (2013). "Environmental scanning electron microscopy gold immunolabeling in cell biology". Cell Imaging Techniques. Methods in Molecular Biology. 931. pp. 517–23. doi:10.1007/978-1-62703-056-4_27. ISBN 978-1-62703-055-7. PMID 23027021.
- Narayanan BK, Kovarik L, Quintana MA, Mills MJ (2013). "Characterisation of ferritic weld microstructures using various electron microscopy techniques: a review". Science and Technology of Welding and Joining. 16 (1): 12–22. doi:10.1179/136217110X12720264008312. ISSN 1362-1718. S2CID 135581795.
- Faulk WP, Taylor GM (November 1971). "An immunocolloid method for the electron microscope". Immunochemistry. 8 (11): 1081–3. doi:10.1016/0019-2791(71)90496-4. PMID 4110101.
- Owen GR, Meredith DO, Ap Gwynn I, Richards RG (2001). "Enhancement of immunogold-labelled focal adhesion sites in fibroblasts cultured on metal substrates: problems and solutions". Cell Biol. Int. 25 (12): 1251–9. doi:10.1006/cbir.2001.0846. PMID 11748918.
External links
| Library resources about Immunolabelling |
- Labeling Procedures
- Molecular cross-talk between the transcription, translation, and nonsense-mediated decay machineries
- Nanoprobes Technical Help: Successful EM Immunolabeling
- Immunolabeling as a tool for understanding the spatial distribution of fiber wall components and their biosynthetic enzymes
{kind=link}