Insulin signal transduction pathway
The insulin transduction pathway is a biochemical pathway by which insulin increases the uptake of glucose into fat and muscle cells and reduces the synthesis of glucose in the liver and hence is involved in maintaining glucose homeostasis. This pathway is also influenced by fed versus fasting states, stress levels, and a variety of other hormones.[1]
When carbohydrates are consumed, digested, and absorbed the pancreas senses the subsequent rise in blood glucose concentration and releases insulin to promote uptake of glucose from the bloodstream. When insulin binds to the insulin receptor, it leads to a cascade of cellular processes that promote the usage or, in some cases, the storage of glucose in the cell. The effects of insulin vary depending on the tissue involved, e.g., insulin is most important in the uptake of glucose by muscle and adipose tissue.[2]
This insulin signal transduction pathway is composed of trigger mechanisms (e.g., autophosphorylation mechanisms) that serve as signals throughout the cell. There is also a counter mechanism in the body to stop the secretion of insulin beyond a certain limit. Namely, those counter-regulatory mechanisms are glucagon and epinephrine. The process of the regulation of blood glucose (also known as glucose homeostasis) also exhibits oscillatory behavior.
On a pathological basis, this topic is crucial to understanding certain disorders in the body such as diabetes, hyperglycemia and hypoglycemia.
Transduction pathway
The functioning of a signal transduction pathway is based on extra-cellular signaling that in turn creates a response that causes other subsequent responses, hence creating a chain reaction, or cascade. During the course of signaling, the cell uses each response for accomplishing some kind of a purpose along the way. Insulin secretion mechanism is a common example of signal transduction pathway mechanism.
Insulin is produced by the pancreas in a region called Islets of Langerhans. In the islets of Langerhans, there are beta-cells, which are responsible for production and storage of insulin. Insulin is secreted as a response mechanism for counteracting the increasing excess amounts of glucose in the blood.
Glucose in the body increases after food consumption. This is primarily due to carbohydrate intake, but to a much lesser degree protein intake ()(). Depending on the tissue type, the glucose enters the cell through facilitated diffusion or active transport. In muscle and adipose tissue, glucose enters through GLUT 4 receptors via facilitated diffusion (). In brain, retina, kidney, RBC, placenta and many other organs, glucose enters using GLUT 1 and GLUT 3. In the beta-cells of the pancreas and in liver cells, glucose enters through the GLUT 2 receptors [3] (process described below).
Insulin biosynthesis and transcription
Insulin biosynthesis is regulated by transcriptional and translational levels. The β-cells promote their protein transcription in response to nutrients. The exposure of rat Langerhans islets to glucose for 1 hour is able to remarkably induce the intracellular proinsulin levels. It was noted that the proinsulin mRNA remained stable. This suggests that the acute response to glucose of the insulin synthesis is independent of mRNA synthesis in the first 45 minutes because the blockage of the transcription decelerated the insulin accumulation during that time.[4] PTBPs, also called Polypyrimidine tract binding proteins, are proteins that regulate the translation of mRNA. They increase the viability of mRNA and provoke the initiation of the translation. PTBP1 enable the insulin gene-specific activation and insulin granule protein mRNA by glucose.[4]
Two aspects of the transduction pathway process are explained below: insulin secretion and insulin action on the cell.

Insulin secretion
The glucose that goes into the bloodstream after food consumption also enters the beta cells in the Islets of Langerhans in the pancreas. The glucose diffuses in the beta-cell facilitated by a GLUT-2 vesicle. Inside the beta cell, the following process occurs:
Glucose gets converted to Glucose-6-Phosphate (G6P) through Glucokinase, and G6P is subsequently oxidized to form ATP. This process inhibits the ATP-sensitive potassium ion channels of the cell causing the Potassium ion channel to close and not function anymore. The closure of the ATP-sensitive potassium channels causes depolarization of the cell membrane causing the cell membrane to stretch which causes the voltage-gated calcium channel on the membrane to open causing an influx of Ca2+ ions. This influx then stimulates fusion of the insulin vesicles to the cell membrane and secretion of insulin in the extracellular fluid outside the beta cell; thus making it enter the bloodstream. [Also Illustrated in Figure 1.1.1].[5]
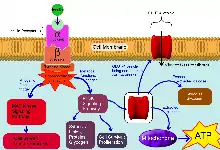
There are 3 subfamilies of Ca+2 channels; L-type Ca+2 channels, Non-L-Type Ca+2 channels (including R-Type) and the T-type Ca+2 channels. There are two phases of the insulin secretion, the first phase involves the L-type Ca+2 channels and the second phase involves the R-type Ca+2 channels. The Ca+2 influx generated by R-type Ca+2 channels is not enough to cause insulin exocytosis, however, it increases the mobilization of the vesicles towards the cell membrane.[4]
Fatty acids and insulin secretion
Fatty acids also affect insulin secretion. In type 2 diabetes, fatty acids are able to potentiate insulin release to compensate the increment need of insulin. It was found that the β-cells express free fatty acid receptors at their surface, through which fatty acids can impact the function of β-cells. Long-chain acyl-CoA and DAG are the metabolites resulting from the intracellular metabolism of fatty acids. Long-chain acyl-CoA has the ability to acylate proteins that are essential in the insulin granule fusion. On the other hand, DAG activates PKC that is involved in the insulin secretion.[4]
Hormonal regulation of insulin secretion
Several hormones can affect insulin secretion. Estrogen is correlated with an increase of insulin secretion by depolarizing the β-cells membrane and enhancing the entry of Ca+2. In contrast, growth hormone is known to lower the serum level of insulin by promoting the production of insulin-like growth factor-I (IGF-I). IGF-I, in turn, suppresses the insulin secretion.[4]
Action on the cell
After insulin enters the bloodstream, it binds to a membrane-spanning glycoprotein receptor. This glycoprotein is embedded in the cellular membrane and has an extracellular receptor domain, made up of two α-subunits, and an intracellular catalytic domain made up of two β-subunits. The α-subunits act as insulin receptors and the insulin molecule acts as a ligand. Together, they form a receptor-ligand complex.
Binding of insulin to the α-subunit results in a conformational change in the membrane-bound glycoprotein, which activates tyrosine kinase domains on each β-subunit. The tyrosine kinase activity causes an auto phosphorylation of several tyrosine residues in the β-subunit. The phosphorylation of 3 residues of tyrosine is necessary for the amplification of the kinase activity.[6]
Once the tyrosine kinase is activated in the insulin receptor, it triggers the activation of the docking proteins, also called IRS (1-4) that are important in the signaling pathway, and then the activation of the PI-3k[7]
The two enzymes Mitogen-activated Protein Kinase (MAP-Kinase) and Phosphatidylinositol-3-Kinase (PI-3K, Phosphoinositide 3-kinase) are responsible for expressing the mitogenic and metabolic actions of Insulin, respectively.
The activation of MAP-Kinase leads to the completion of mitogenic functions like cell growth and gene expression.
The activation of PI-3K leads to crucial metabolic functions such as synthesis of lipids, proteins and glycogen. It also leads to cell survival and cell proliferation. Most importantly, the PI-3K pathway is responsible for the distribution of glucose for important cell functions. The activation of PI-3K leads to the activation of PKB (AKT) that induces the impact of insulin on the liver. For example, the suppression of hepatic glucose synthesis and the activation of glycogen synthesis. Hence, PKB possesses a crucial role in the linkage of the glucose transporter (GLUT4) to the insulin signaling pathway. The activated GLUT4 will translocate to the cell membrane and promotes the transportation of glucose into the intracellular medium.[6]
Thus, insulin's role is more of a promoter for the usage of glucose in the cells rather than neutralizing or counteracting it.
Regulation of the insulin receptor signal
PI-3K is one of the important components in the regulation of the insulin signaling pathway. It maintains the insulin sensitivity in the liver. PI-3K is composed of a regulatory subunit (P85) and a catalytic subunit (P110). P85 regulates the activation of PI-3K enzyme.[8] In the PI-3K heterodimer (P85-p110), P85 is responsible for the PI-3K activity, by binding to the binding site on the insulin receptor substrates (IRS). It was noted that an increase of P85 a (isoform of P85) results in a competition between the later and the P85-P110 complex to the IRS binding site, reducing the PI-3k activity and leading to insulin resistance. Insulin resistance refers also to Type 2 diabetes. It was also noted that increased serine phosphorylation of IRS is involved in the insulin resistance by reducing their ability to attract PI3K. The serine phosphorylation can also lead to degradation of IRS-1.[7]
Feedback mechanisms
Signal transduction is a mechanism in which the cell responds to a signal from the environment by activating several proteins and enzymes that will give a response to the signal. Feedback mechanism might involve negative and positive feedbacks. In the negative feedback, the pathway is inhibited and the final result of the transduction pathway is reduced or limited. In positive feedback, the transduction pathway is promoted and stimulated to produce more products.
Positive
Insulin secretion results in positive feedback in different ways. Firstly, insulin increases the uptake of glucose from blood by the translocation and exocytosis of GLUT4 storage vesicles in the muscle and fat cells. Secondly, it promotes the conversion of glucose into triglyceride in the liver, fat, and muscle cells. Finally, the cell will increase the rate of glycolysis within itself to break glucose in the cell into other components for tissue growth purposes.
An example of positive feedback mechanism in the insulin transduction pathway is the activation of some enzymes that inhibit other enzymes from slowing or stopping the insulin transduction pathway which results in improved intake of the glucose.
One of these pathways, involves the PI(3)K enzyme (Phosphoinositide 3-kinase). This pathway is responsible for activating glycogen, lipid-protein synthesis, and specific gene expression of some proteins which will help in the intake of glucose. Different enzymes control this pathway. Some of these enzymes constrict the pathway causing a negative feedback like the GSK-3 pathway. Other enzymes will push the pathway forward causing a positive feedback like the AKT and P70 enzymes. When insulin binds to its receptor, it activates the glycogen synthesis by inhibiting the enzymes that slow down the PI(3)K pathway such as PKA enzyme. At the same time, it will promote the function of the enzymes that provide a positive feedback for the pathway like the AKT and P70 enzymes.[9] The inactivation of the enzymes that stop the reaction and activating of enzymes that provide a positive feedback will increase glycogen, lipid & protein syntheses and promote glucose intake.
(Image to help explain the function of the proteins mentioned above in the positive feedback.)
Negative
When insulin binds to the cell's receptor, it results in negative feedback by limiting or stopping some other actions in the cell. It inhibits the release and production of glucose from the cells which is an important part in reducing the glucose blood level. Insulin will also inhibit the breakdown of glycogen into glucose by inhibiting the expression of the enzymes that catalyzes the degradation of Glycogen.
An example of negative feedback is slowing or stopping the intake of glucose after the pathway was activated. Negative feedback is shown in the insulin signal transduction pathway by constricting the phosphorylation of the insulin-stimulated tyrosine.[10] The enzyme that deactivates or phosphorylates the insulin-stimulated tyrosine is called tyrosine phosphatases (PTPases). When activated, this enzyme provides a negative feedback by catalyzing the dephosphorylation of the insulin receptors.[11] The dephosphorylation of the insulin receptor slows down glucose intake by inhibiting the activation (phosphorylation) of proteins responsible for further steps of the insulin transduction pathway.
Trigger mechanism
Insulin is synthesized and secreted in the beta cells of the islets of Langerhans. Once insulin is synthesized, the beta cells are ready to release it in two different phases. As for the first phase, insulin release is triggered rapidly when the blood glucose level is increased. The second phase is a slow release of newly formed vesicles that are triggered regardless of the blood sugar level. Glucose enters the beta cells and goes through glycolysis to form ATP that eventually causes depolarization of the beta cell membrane (as explained in Insulin secretion section of this article). The depolarization process causes voltage-controlled calcium channels (Ca2+) opening, allowing the calcium to flow into the cells. An increased calcium level activates phospholipase C, which cleaves the membrane phospholipid phosphatidylinositol 4,5-bisphosphate into Inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 binds to receptor proteins in the membrane of the endoplasmic reticulum (ER). This releases (Ca2+) from the ER via IP3 gated channels, and raises the cell concentration of calcium even more. The influx of Ca2+ ions causes the secretion of insulin stored in vesicles through the cell membrane. The process of insulin secretion is an example of a trigger mechanism in a signal transduction pathway because insulin is secreted after glucose enters the beta cell and that triggers several other processes in a chain reaction.
Counter mechanism
Function of glucagon
While insulin is secreted by the pancreas to lower blood glucose levels, glucagon is secreted to raise blood glucose levels. This is why glucagon has been known for decades as a counter-regulatory hormone.[12] When blood glucose levels are low, the pancreas secretes glucagon, which in turn causes the liver to convert stored glycogen polymers into glucose monomers, which is then released into the blood. This process is called glycogenolysis. Liver cells, or hepatocytes, have glucagon receptors which allow for glucagon to attach to them and thus stimulate glycogenolysis.[13] Contrary to insulin, which is produced by pancreatic β-cells, glucagon is produced by pancreatic α-cells.[14] It is also known that an increase in insulin suppresses glucagon secretion, and a decrease in insulin, along with low glucose levels, stimulates the secretion of glucagon.[14]
Oscillatory behavior
When blood glucose levels are too low, the pancreas is signaled to release glucagon, which has essentially the opposite effect of insulin and therefore opposes the reduction of glucose in the blood. Glucagon is delivered directly to the liver, where it connects to the glucagon receptors on the membranes of the liver cells, signals the conversion of the glycogen already stored in the liver cells into glucose. This process is called glycogenolysis.
Conversely, when the blood glucose levels are too high, the pancreas is signaled to release insulin. Insulin is delivered to the liver and other tissues throughout the body (e.g., muscle, adipose). When the insulin is introduced to the liver, it connects to the insulin receptors already present, that is tyrosine kinase receptor.[15] These receptors have two alpha subunits (extracellular) and two beta subunits (intercellular) which are connected through the cell membrane via disulfide bonds. When the insulin binds to these alpha subunits, 'glucose transport 4' (GLUT4) is released and transferred to the cell membrane to regulate glucose transport in and out of the cell. With the release of GLUT4, the allowance of glucose into cells is increased, and therefore the concentration of blood glucose might decrease. This, in other words, increases the utilization of the glucose already present in the liver. This is shown in the adjacent image. As glucose increases, the production of insulin increases, which thereby increases the utilization of the glucose, which maintains the glucose levels in an efficient manner and creates an oscillatory behavior.
References
- Rhoads, Robert E. (17 July 2001). Signaling Pathways for Translation: Insulin and Nutrients. Springer Science & Business Media. ISBN 978-3-540-41709-5.
- Srivastava, Ashok K.; Posner, Barry I. (6 December 2012). Insulin Action. Springer Science & Business Media. ISBN 978-1-4615-5647-3.
- Ganong WF (2016). "Chapter 24: Endocrine functions of the pancreas and regulation of carbohydrate metabolism". Review of medical physiology (25th ed.). New Delhi: McGraw Hill. pp. 432–433. ISBN 978-93-392-2328-1.
- Fu, Zhuo; Gilbert, Elizabeth R.; Liu, Dongmin (1 January 2013). "Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes". Current Diabetes Reviews. 9 (1): 25–53. doi:10.2174/157339913804143225. PMC 3934755. PMID 22974359.
- Guyton AC, Hall JE (2005). "Chapter 78: Insulin, Glucagon, and Diabetes Mellitus". Textbook of medical physiology (11th ed.). Philadelphia: W.B. Saunders. pp. 963–68. ISBN 978-0-7216-0240-0.
- Saini, Vandana (15 July 2010). "Molecular mechanisms of insulin resistance in type 2 diabetes mellitus". World Journal of Diabetes. 1 (3): 68–75. doi:10.4239/wjd.v1.i3.68. PMC 3083885. PMID 21537430.
- Draznin, Boris (2006). "Molecular Mechanisms of Insulin Resistance: Serine Phosphorylation of Insulin Receptor Substrate-1 and Increased Expression of p85α" (PDF). American Diabetes Association. Retrieved 29 October 2017.
- Taniguchi, Cullen M.; Tran, Thien T.; Kondo, Tatsuya; Luo, Ji; Ueki, Kohjiro; Cantley, Lewis C.; Kahn, C. Ronald (8 August 2006). "Phosphoinositide 3-kinase regulatory subunit p85 suppresses insulin action via positive regulation of PTEN". PNAS. 103 (32): 12093–12097. doi:10.1073/pnas.0604628103. PMC 1524929. PMID 16880400.
- Brady MJ, Nairn AC, Saltiel AR (November 1997). "The regulation of glycogen synthase by protein phosphatase 1 in 3T3-L1 adipocytes. Evidence for a potential role for DARPP-32 in insulin action". The Journal of Biological Chemistry. 272 (47): 29698–703. doi:10.1074/jbc.272.47.29698. PMID 9368038.
- Craparo A, Freund R, Gustafson TA (April 1997). "14-3-3 (epsilon) interacts with the insulin-like growth factor I receptor and insulin receptor substrate I in a phosphoserine-dependent manner". The Journal of Biological Chemistry. 272 (17): 11663–9. doi:10.1074/jbc.272.17.11663. PMID 9111084.
- Saltiel AR, Kahn CR (December 2001). "Insulin signalling and the regulation of glucose and lipid metabolism" (PDF). Nature. 414 (6865): 799–806. Bibcode:2001Natur.414..799S. doi:10.1038/414799a. hdl:2027.42/62568. PMID 11742412. S2CID 1119157.
- Brubaker PL, Anini Y (November 2003). "Direct and indirect mechanisms regulating secretion of glucagon-like peptide-1 and glucagon-like peptide-2". Canadian Journal of Physiology and Pharmacology. 81 (11): 1005–12. doi:10.1139/y03-107. PMID 14719035.
- Estall JL, Drucker DJ (May 2006). "Glucagon and glucagon-like peptide receptors as drug targets". Current Pharmaceutical Design. 12 (14): 1731–50. doi:10.2174/138161206776873671. PMID 16712485.
- Cooperberg BA, Cryer PE (November 2010). "Insulin reciprocally regulates glucagon secretion in humans". Diabetes. 59 (11): 2936–40. doi:10.2337/db10-0728. PMC 2963553. PMID 20811038.
- Khan AH, Pessin JE (November 2002). "Insulin regulation of glucose uptake: a complex interplay of intracellular signalling pathways". Diabetologia. 45 (11): 1475–83. doi:10.1007/s00125-002-0974-7. PMID 12436329.CS1 maint: uses authors parameter (link)