Localized molecular orbitals
Localized molecular orbitals are molecular orbitals which are concentrated in a limited spatial region of a molecule, such as a specific bond or lone pair on a specific atom. They can be used to relate molecular orbital calculations to simple bonding theories, and also to speed up post-Hartree–Fock electronic structure calculations by taking advantage of the local nature of electron correlation. Localized orbitals in systems with periodic boundary conditions are known as Wannier functions.
Standard ab initio quantum chemistry methods lead to delocalized orbitals that, in general, extend over an entire molecule and have the symmetry of the molecule. Localized orbitals may then be found as linear combinations of the delocalized orbitals, given by an appropriate unitary transformation.
In the water molecule for example, ab initio calculations show bonding character primarily in two molecular orbitals, each with electron density equally distributed among the two O-H bonds. The localized orbital corresponding to one O-H bond is the sum of these two delocalized orbitals, and the localized orbital for the other O-H bond is their difference; as per Valence bond theory.
For multiple bonds and lone pairs, different localization procedures give different orbitals. The Boys and Edmiston-Ruedenberg localization methods mix these orbitals to give equivalent bent bonds in ethylene and rabbit ear lone pairs in water, while the Pipek-Mezey method preserves their respective σ and π symmetry.
Equivalence of localized and delocalized orbital descriptions
For molecules with a closed electron shell, in which each molecular orbital is doubly occupied, the localized and delocalized orbital descriptions are in fact equivalent and represent the same physical state. It might seem, again using the example of water, that placing two electrons in the first bond and two other electrons in the second bond is not the same as having four electrons free to move over both bonds. However, in quantum mechanics all electrons are identical and cannot be distinguished as same or other. The total wavefunction must have a form which satisfies the Pauli exclusion principle such as a Slater determinant (or linear combination of Slater determinants), and it can be shown [1] that if two electrons are exchanged, such a function is unchanged by any unitary transformation of the doubly occupied orbitals.
For molecules with an open electron shell, in which some molecular orbitals are singly occupied, the electrons of alpha and beta spin must be localized separately.[2][3] This applies to radical species such as nitric oxide and dioxygen. Again, in this case the localized and delocalized orbital descriptions are equivalent and represent the same physical state.
Computation methods
Localized molecular orbitals (LMO)[4] are obtained by unitary transformation upon a set of canonical molecular orbitals (CMO). The transformation usually involves the optimization (either minimization or maximization) of the expectation value of a specific operator. The generic form of the localization potential is:
,

where is the localization operator and is a molecular spatial orbital. Many methodologies have been developed during the past decades, differing in the form of .


The optimization of the objective function is usually performed using pairwise Jacobi rotations.[5] However, this approach is prone to saddle point convergence (if it even converges), and thus other approaches have also been developed, from simple conjugate gradient methods with exact line searches,[6] to Newton-Raphson[7] and trust-region methods.[8]
Foster-Boys
The Foster-Boys (also known as Boys) localization method[9] minimizes the spatial extent of the orbitals by minimizing , where . This turns out to be equivalent[10][11] to the easier task of maximizing . In one dimension, the Foster-Boys (FB) objective function can also be written as



.[12]

Fourth moment
The fourth moment (FM) procedure[12] is analogous to Foster-Boys scheme, however the orbital fourth moment is used instead of the orbital second moment. The objective function to be minimized is
.

The fourth moment method produces more localized virtual orbitals than Foster-Boys method,[12] since it implies a larger penalty on the delocalized tails. For graphene (a delocalized system), the fourth moment method produces more localized occupied orbitals than Foster-Boys and Pipek-Mezey schemes.[12]
Edmiston-Ruedenberg
Edmiston-Ruedenberg localization[5] maximizes the electronic self-repulsion energy by maximizing , where .


Pipek-Mezey
Pipek-Mezey localization[13] takes a slightly different approach, maximizing the sum of orbital-dependent partial charges on the nuclei:
.

Pipek and Mezey originally used Mulliken charges, which are mathematically ill defined. Recently, Pipek-Mezey style schemes based on a variety of mathematically well-defined partial charge estimates have been discussed.[14] Some notable choices are Voronoi charges,[14] Becke charges,[14] Hirshfeld or Stockholder charges,[14] intrinsic atomic orbital charges,[15] Bader charges,[16] or "fuzzy atom" charges.[17] Rather surprisingly, despite of the wide variation in the (total) partial charges reproduced by the different estimates, analysis of the resulting Pipek-Mezey orbitals has shown that the localized orbitals are rather insensitive to the partial charge estimation scheme used in the localization process.[14] However, due to the ill-defined mathematical nature of Mulliken charges (and Löwdin charges, which have also been used in some works[18]), as better alternatives are nowadays available it is advisable to use them in favor of the original version.
The most important quality of the Pipek-Mezey scheme is that it preserves σ-π separation in planar systems, which sets it apart from the Foster-Boys and Edmiston-Ruedenberg schemes that mix σ and π bonds. This property holds independent of the partial charge estimate used.[13][14][15][16][17]
While the usual formulation of the Pipek-Mezey method invokes an iterative procedure to localize the orbitals, a non-iterative method has also been recently suggested.[19]
In organic chemistry
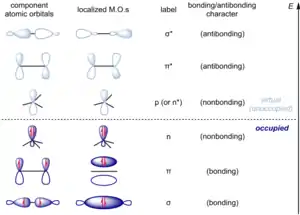
Organic chemistry is often discussed in terms of localized molecular orbitals in a qualitative and informal sense. Historically, much of classical organic chemistry was built on the older valence bond / orbital hybridization models of bonding. To account for phenomena like aromaticity, this simple model of bonding is supplemented by semi-quantitative results from Hückel molecular orbital theory. However, the understanding of stereoelectronic effects requires the analysis of interactions between donor and acceptor orbitals between two molecules or different regions within the same molecule, and molecular orbitals must be considered. Because proper (symmetry-adapted) molecular orbitals are fully delocalized and do not admit a ready correspondence with the "bonds" of the molecule, as visualized by the practicing chemist, the most common approach is to instead consider the interaction between filled and unfilled localized molecular orbitals that correspond to σ bonds, π bonds, lone pairs, and their unoccupied counterparts. These orbitals and typically given the notation σ (sigma bonding), π (pi bonding), n (occupied nonbonding orbital, "lone pair"), p (unoccupied nonbonding orbital, "empty p orbital"; the symbol n* for unoccupied nonbonding orbital is seldom used), π* (pi antibonding), and σ* (sigma antibonding). (Woodward and Hoffmann use ω for nonbonding orbitals in general, occupied or unoccupied.) When comparing localized molecular orbitals derived from the same atomic orbitals, these classes generally follow the order σ < π < n < p (n*) < π* < σ* when ranked by increasing energy. [20]
The localized molecular orbitals that organic chemists often depict can be thought of as qualitative renderings of orbitals generated by the computational methods described above. However, they do not map onto any single approach, nor are they used consistently. For instance, the lone pairs of water are usually treated as two equivalent spx hybrid orbitals, while the corresponding "nonbonding" orbitals of carbenes are generally treated as a filled σ(out) orbital and an unfilled pure p orbital, even though the lone pairs of water could be described analogously by filled σ(out) and p orbitals (for further discussion, see the article on lone pair and the discussion above on sigma-pi and equivalent-orbital models). In other words, the type of localized orbital invoked depends on context and considerations of convenience and utility.
References
- Levine I.N., “Quantum Chemistry” (4th ed., Prentice-Hall 1991) sec.15.8
- Hirst, D. M.; Linington, Mary E. (1970). "Localized orbitals for the oxygen and nitric oxide molecules". Theoretica Chimica Acta. 16 (1): 55–62. doi:10.1007/BF01045967. S2CID 95235964.
- Duke, Brian J. (1987). "Linnett's double quartet theory and localised orbitals". Journal of Molecular Structure: THEOCHEM. 152 (3–4): 319–330. doi:10.1016/0166-1280(87)80072-6.
- Jensen, Frank (2007). Introduction to Computational Chemistry. Chichester, England: John Wiley and Sons. pp. 304–308. ISBN 978-0-470-01187-4.
- Edmiston, Clyde; Ruedenberg, Klaus (1963). "Localized Atomic and Molecular Orbitals". Reviews of Modern Physics. 35 (3): 457–465. Bibcode:1963RvMP...35..457E. doi:10.1103/RevModPhys.35.457.
- Lehtola, Susi; Jónsson, Hannes (2013). "Unitary Optimization of Localized Molecular Orbitals". Journal of Chemical Theory and Computation. 9 (12): 5365–5372. doi:10.1021/ct400793q. PMID 26592274.
- Leonard, Joseph M.; Luken, William L. (1982). "Quadratically Convergent Calculation of Localized Molecular Orbitals". Theoretica Chimica Acta. 62 (2): 107–132. doi:10.1007/BF00581477. S2CID 97499582.
- Høyvik, Ida-Marie; Jansik, Branislav; Jørgensen, Poul (2012). "Trust Region Minimization of Orbital Localization Functions". Journal of Chemical Theory and Computation. 8 (9): 3137–3146. doi:10.1021/ct300473g. PMID 26605725.
- Boys, S. F. (1960). "Construction of Molecular orbitals to be minimally variant for changes from one molecule to another". Reviews of Modern Physics. 32 (2): 296–299. Bibcode:1960RvMP...32..296B. doi:10.1103/RevModPhys.32.300.
- Kleier, Daniel; J. Chem. Phys. 61, 3905 (1974) (1974). "Localized molecular orbitals for polyatomic molecules. I. A comparison of the Edmiston-Ruedenberg and Boys localization methods". The Journal of Chemical Physics. Journal of Chemical Physics. 61 (10): 3905–3919. Bibcode:1974JChPh..61.3905K. doi:10.1063/1.1681683.
- Introduction to Computational Chemistry by Frank Jensen 1999, page 228 equation 9.27
- Høyvik, Ida-Marie; Jansik, Branislav; Jørgensen, Poul (2012). "Orbital localization using fourth central moment minimization" (PDF). Journal of Chemical Physics. 137 (22): 244114. Bibcode:2012JChPh.137v4114H. doi:10.1063/1.4769866. PMID 23248994.
- Pipek, János; Mezey, Paul G. (1989). "A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions". The Journal of Chemical Physics. 90 (9): 4916. Bibcode:1989JChPh..90.4916P. doi:10.1063/1.456588.
- Lehtola, Susi; Jónsson, Hannes (8 January 2014). "Pipek–Mezey orbital localization using various partial charge estimates". Journal of Chemical Theory and Computation. 10 (2): 642–649. doi:10.1021/ct401016x. PMID 26580041.
- Knizia, G. (2013). "Intrinsic Atomic Orbitals: An Unbiased Bridge between Quantum Theory and Chemical Concepts". Journal of Chemical Theory and Computation. 9 (11): 4834–4843. arXiv:1306.6884. Bibcode:2013arXiv1306.6884K. doi:10.1021/ct400687b. PMID 26583402. S2CID 17717923.
- Cioslowski, J. (1991). "Partitioning of the orbital overlap matrix and the localization criteria". Journal of Mathematical Chemistry. 8 (1): 169–178. doi:10.1007/BF01166933. S2CID 96731740.
- Alcoba, Diego R.; Lain, Luis; Torre, Alicia; Bochicchio, Roberto C. (15 April 2006). "An orbital localization criterion based on the theory of "fuzzy" atoms". Journal of Computational Chemistry. 27 (5): 596–608. doi:10.1002/jcc.20373. PMID 16470667.
- Høyvik, Ida-Marie; Jansik, Branislav; Jørgensen, Poul (3 April 2013). "Pipek–Mezey localization of occupied and virtual orbitals". Journal of Computational Chemistry. 34 (17): 1456–1462. doi:10.1002/jcc.23281. PMID 23553349.
- Heßelmann, Andreas (10 May 2016). "Local Molecular Orbitals from a Projection onto Localized Centers". Journal of Chemical Theory and Computation. 12 (6): 2720–2741. doi:10.1021/acs.jctc.6b00321. PMID 27164445.
- Kirby, A. J. (2002). Stereoelectronic Effects. Oxford, UK: Oxford University Press. ISBN 978-0198558934.