Matched molecular pair analysis
Matched molecular pair analysis (MMPA) is a method in cheminformatics that compares the properties of two molecules that differ only by a single chemical transformation, such as the substitution of a hydrogen atom by a chlorine one. Such pairs of compounds are known as matched molecular pairs (MMP). Because the structural difference between the two molecules is small, any experimentally observed change in a physical or biological property between the matched molecular pair can more easily be interpreted. The term was first coined by Kenny and Sadowski in the book Chemoinformatics in Drug Discovery.[1]
Introduction
MMP can be defined as a pair of molecules that differ in only a minor single point change (See Fig 1). Matched molecular pairs (MMPs) are widely used in medicinal chemistry to study changes in compound properties which includes biological activity, toxicity, environmental hazards and much more, which are associated with well-defined structural modifications. Single point changes in the molecule pairs are termed a chemical transformation or Molecular transformation. Each molecular pair is associated with a particular transformation. An example of transformation is the replacement of one functional group by another. More specifically, molecular transformation can be defined as the replacement of a molecular fragment having one, two or three attachment points with another fragment. Useful Molecular transformation in a specified context is termed as "Significant" transformations. For example, a transformation may systematically decrease or increase a desired property of chemical compounds. Transformations that affect a particular property/activity in a statistically significant sense are called as significant transformations. The transformation is considered significant, if it increases the property value "more often" than it decreases it or vice versa. Thus, the distribution of increasing and decreasing pairs should be significantly different from the binomial ("no effect") distribution with a particular p-value (usually 0.05).
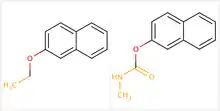
Significance of MMP based analysis
MMP based analysis is an attractive method for computational analysis because they can be algorithmically generated and they make it possible to associate defined structural modifications at the level of compound pairs with chemical property changes, including biological activity.[2][3][4]
Interpretable QSAR models
MMPA is quite useful in the field of quantitative structure–activity relationship (QSAR) modelling studies. One of the issues of QSAR models is they are difficult to interpret in a chemically meaningful manner. While it can be pretty easy to interpret simple linear regression models, the most powerful algorithms like neural networks, support vector machine are similar to "black boxes", which provide predictions that can't be easily interpreted.[5] This problem undermines the applicability of QSAR model in helping the medicinal chemist to make the decision. If the compound is predicted to be active against some microorganism, what are the driving factors of its activity? Or if it is predicted to be inactive, how its activity can be modulated? The black box nature of the QSAR model prevents it from addressing these crucial issues. The use of predicted MMPs allows to interpret models and identify which MMPs were learned by the model.[6] The MMPs, which were not reproduced by the model, could correspond to experimental errors or deficiency of the model (inappropriate descriptors, too few data, etc.).
Analysis of MMPs (matched molecular pair) can be very useful for understanding the mechanism of action. A medicinal chemist might be interested particularly in "activity cliff". Activity cliff is a minor structural modification, which changes the target activity significantly.
Activity Cliff
Activity cliffs are minor structural modifications, with significant effect on molecular property. Activity cliffs usually have high SAR information content. Because small chemical changes in the set of similar compounds lead to large changes in activity. The assessment of activity cliffs requires careful consideration of similarity and potency difference criteria[7][8][9]
Types of MMP based analysis
Matched molecular pair (MMPA) analyses can be classified into two types: supervised and unsupervised MMPA.
Supervised MMPA
In supervised MMPA, the chemical transformations are predefined, then the corresponding matched pair compounds are found within the data set and the change in end point computed for each transformation.
Unsupervised MMPA
Also known as automated MMPAs. A machine learning algorithm is used to finds all possible matched pairs in a data set according to a set of predefined rules. This results in much larger numbers of matched pairs and unique transformations, which are typically filtered during the process to identify those transformations that correspond to statistically significant changes in the targeted property with a reasonable number of matched pairs.
Matched molecular series
Here instead of looking at the pair of molecules which differ only at one point, a series of more than 2 molecules different at a single point is considered. The concept of matching molecular series was introduced by Wawer and Bajorath.[10] It is argued that longer matched series is more likely to exhibit preferred molecular transformation while, matched pairs exhibit only a small preference.[11]
Limitations
The application of the MMPA across large chemical databases for the optimization of ligand potency is problematic because same structural transformation may increase or decrease or doesn't affect the potency of different compounds in the dataset. Selection of practical significant transformation from a dataset of molecules is a challenging issue in the MMPA. Moreover, the effect of a particular molecular transformation can significantly depend on the Chemical context of transformations.[12][13]
Beside these, MMPA might pose some limitations in terms of computational resources, especially when dealing with databases of compounds with a large number of breakable bonds. Further, more atoms in the variable part of the molecule also leads to combinatorial explosion problems. To deal with this, the number of breakable bonds and number of atoms in the variable part can be used to pre-filter the database.
References
- Kenny, Peter W.; Sadowski, Jens (2005). "Chapter 11: Structure Modification in Chemical Databases". In Oprea, Tudor I. (ed.). Chemoinformatics in Drug Discovery. Wiley-VCH Verlag GmbH & Co. KGaA. pp. 271–285.
- Griffen, Ed; Leach, Andrew G.; Robb, Graeme R.; Warner, Daniel J. (2011). "Matched molecular pairs as a medicinal chemistry tool". J. Med. Chem. 54 (22): 7739–50. doi:10.1021/jm200452d. PMID 21936582.
- Wassermann, A.M.; Dimova, D.; Iyer P; et al. (2012). "Advances in computational medicinal chemistry: matched molecular pair analysis". Drug Development Research. 73 (8): 518–527. doi:10.1002/ddr.21045.
- Dossetter, Alexander G.; Griffen, Edward J.; Leach, Andrew G. (2013). "Matched molecular pair analysis in drug discovery". Drug Discovery Today. 18 (15–16): 724–731. doi:10.1016/j.drudis.2013.03.003. PMID 23557664.
- Cumming et.al., Chemical predictive modelling to improve compound quality,Nature Reviews Drug Discovery 12, 948–962 (2013)
- Sushko, Yurii; Novotarskyi, Sergii; Körner, Robert; Vogt, Joachim; Abdelaziz, Ahmed; Tetko, Igor V (2014-12-11). "Prediction-driven matched molecular pairs to interpret QSARs and aid the molecular optimization process". Journal of Cheminformatics. 6 (1): 48. doi:10.1186/s13321-014-0048-0. PMC 4272757. PMID 25544551.
- Stumpfe D, Bajorath J: Exploring activity cliffs in medicinal chemistry. J Med Chem. 2012; 55(7): 2932–2942
- Stumpfe D, Hu Y, Dimova D, et al.: Recent progress in understanding activity cliffs and their utility in medicinal chemistry. J Med Chem. 2014; 57(1): 18–28.
- Hu Y, Stumpfe D, Bajorath J: Advancing the activity cliff concept [v1; ref status: indexed, http://f1000r.es/1wf]. F1000Res. 2013; 2: 199.
- Wawer, Mathias; Bajorath, Jürgen (2011). "Local Structural Changes, Global Data Views: Graphical Substructure−Activity Relationship Trailing". J. Med. Chem. 54 (8): 2944–2951. doi:10.1021/jm200026b. PMID 21443196.
- O'Boyle, Noel M.; Boström, Jonas; Sayle, Roger A.; Gill, Adrian (2014). "Using Matched Molecular Series as a Predictive Tool To Optimize Biological Activity". J. Med. Chem. 57 (6): 2704–2713. doi:10.1021/jm500022q. PMC 3968889. PMID 24601597.
- Warner, D. J., Bridgland-Taylor, M. H., Sefton, C. E. & Wood, D. J. Prospective prediction of antitarget activity by matched molecular pairs analysis. Mol. Inform. 31, 365–368 (2012).
- Hajduk, P.J.; Sauer, D.R. (2008). "Statistical analysis of the effects of common chemical substituents on ligand potency". J. Med. Chem. 51 (3): 553–64. doi:10.1021/jm070838y. PMID 18173228.