Nuclear magnetic resonance spectroscopy of proteins
Nuclear magnetic resonance spectroscopy of proteins (usually abbreviated protein NMR) is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins, and also nucleic acids, and their complexes. The field was pioneered by Richard R. Ernst and Kurt Wüthrich at the ETH,[1] and by Ad Bax, Marius Clore, Angela Gronenborn at the NIH,[2] and Gerhard Wagner at Harvard University, among others. Structure determination by NMR spectroscopy usually consists of several phases, each using a separate set of highly specialized techniques. The sample is prepared, measurements are made, interpretive approaches are applied, and a structure is calculated and validated.
NMR involves the quantum-mechanical properties of the central core ("nucleus") of the atom. These properties depend on the local molecular environment, and their measurement provides a map of how the atoms are linked chemically, how close they are in space, and how rapidly they move with respect to each other. These properties are fundamentally the same as those used in the more familiar magnetic resonance imaging (MRI), but the molecular applications use a somewhat different approach, appropriate to the change of scale from millimeters (of interest to radiologists) to nanometers (bonded atoms are typically a fraction of a nanometer apart), a factor of a million. This change of scale requires much higher sensitivity of detection and stability for long term measurement. In contrast to MRI, structural biology studies do not directly generate an image, but rely on complex computer calculations to generate three-dimensional molecular models.
Currently most samples are examined in a solution in water, but methods are being developed to also work with solid samples. Data collection relies on placing the sample inside a powerful magnet, sending radio frequency signals through the sample, and measuring the absorption of those signals. Depending on the environment of atoms within the protein, the nuclei of individual atoms will absorb different frequencies of radio signals. Furthermore, the absorption signals of different nuclei may be perturbed by adjacent nuclei. This information can be used to determine the distance between nuclei. These distances in turn can be used to determine the overall structure of the protein.
A typical study might involve how two proteins interact with each other, possibly with a view to developing small molecules that can be used to probe the normal biology of the interaction ("chemical biology") or to provide possible leads for pharmaceutical use (drug development). Frequently, the interacting pair of proteins may have been identified by studies of human genetics, indicating the interaction can be disrupted by unfavorable mutations, or they may play a key role in the normal biology of a "model" organism like the fruit fly, yeast, the worm C. elegans, or mice. To prepare a sample, methods of molecular biology are typically used to make quantities by bacterial fermentation. This also permits changing the isotopic composition of the molecule, which is desirable because the isotopes behave differently and provide methods for identifying overlapping NMR signals.
Sample preparation

Protein nuclear magnetic resonance is performed on aqueous samples of highly purified protein. Usually, the sample consists of between 300 and 600 microlitres with a protein concentration in the range 0.1 – 3 millimolar. The source of the protein can be either natural or produced in a production system using recombinant DNA techniques through genetic engineering. Recombinantly expressed proteins are usually easier to produce in sufficient quantity, and this method makes isotopic labeling possible.
The purified protein is usually dissolved in a buffer solution and adjusted to the desired solvent conditions. The NMR sample is prepared in a thin-walled glass tube.
Data collection
Protein NMR utilizes multidimensional nuclear magnetic resonance experiments to obtain information about the protein. Ideally, each distinct nucleus in the molecule experiences a distinct electronic environment and thus has a distinct chemical shift by which it can be recognized. However, in large molecules such as proteins the number of resonances can typically be several thousand and a one-dimensional spectrum inevitably has incidental overlaps. Therefore, multidimensional experiments that correlate the frequencies of distinct nuclei are performed. The additional dimensions decrease the chance of overlap and have a larger information content, since they correlate signals from nuclei within a specific part of the molecule. Magnetization is transferred into the sample using pulses of electromagnetic (radiofrequency) energy and between nuclei using delays; the process is described with so-called pulse sequences. Pulse sequences allow the experimenter to investigate and select specific types of connections between nuclei. The array of nuclear magnetic resonance experiments used on proteins fall in two main categories — one where magnetization is transferred through the chemical bonds, and one where the transfer is through space, irrespective of the bonding structure. The first category is used to assign the different chemical shifts to a specific nucleus, and the second is primarily used to generate the distance restraints used in the structure calculation, and in the assignment with unlabelled protein.
Depending on the concentration of the sample, on the magnetic field of the spectrometer, and on the type of experiment, a single multidimensional nuclear magnetic resonance experiment on a protein sample may take hours or even several days to obtain suitable signal-to-noise ratio through signal averaging, and to allow for sufficient evolution of magnetization transfer through the various dimensions of the experiment. Other things being equal, higher-dimensional experiments will take longer than lower-dimensional experiments.
Typically, the first experiment to be measured with an isotope-labelled protein is a 2D heteronuclear single quantum correlation (HSQC) spectrum, where "heteronuclear" refers to nuclei other than 1H. In theory, the heteronuclear single quantum correlation has one peak for each H bound to a heteronucleus. Thus, in the 15N-HSQC, with a 15N labelled protein, one signal is expected for each nitrogen atom in the back bone, with the exception of proline, which has no amide-hydrogen due to the cyclic nature of its backbone. Additional 15N-HSQC signals are contributed by each residue with a nitrogen-hydrogen bond in its side chain (W, N, Q, R, H, K). The 15N-HSQC is often referred to as the fingerprint of a protein because each protein has a unique pattern of signal positions. Analysis of the 15N-HSQC allows researchers to evaluate whether the expected number of peaks is present and thus to identify possible problems due to multiple conformations or sample heterogeneity. The relatively quick heteronuclear single quantum correlation experiment helps determine the feasibility of doing subsequent longer, more expensive, and more elaborate experiments. It is not possible to assign peaks to specific atoms from the heteronuclear single quantum correlation alone.
Resonance assignment
In order to analyze the nuclear magnetic resonance data, it is important to get a resonance assignment for the protein, that is to find out which chemical shift corresponds to which atom. This is typically achieved by sequential walking using information derived from several different types of NMR experiment. The exact procedure depends on whether the protein is isotopically labelled or not, since a lot of the assignment experiments depend on carbon-13 and nitrogen-15.
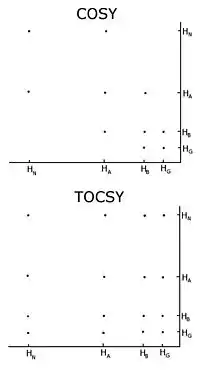
Homonuclear nuclear magnetic resonance
With unlabelled protein the usual procedure is to record a set of two-dimensional homonuclear nuclear magnetic resonance experiments through correlation spectroscopy (COSY), of which several types include conventional correlation spectroscopy, total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY).[3][4] A two-dimensional nuclear magnetic resonance experiment produces a two-dimensional spectrum. The units of both axes are chemical shifts. The COSY and TOCSY transfer magnetization through the chemical bonds between adjacent protons. The conventional correlation spectroscopy experiment is only able to transfer magnetization between protons on adjacent atoms, whereas in the total correlation spectroscopy experiment the protons are able to relay the magnetization, so it is transferred among all the protons that are connected by adjacent atoms. Thus in a conventional correlation spectroscopy, an alpha proton transfers magnetization to the beta protons, the beta protons transfers to the alpha and gamma protons, if any are present, then the gamma proton transfers to the beta and the delta protons, and the process continues. In total correlation spectroscopy, the alpha and all the other protons are able to transfer magnetization to the beta, gamma, delta, epsilon if they are connected by a continuous chain of protons. The continuous chain of protons are the sidechain of the individual amino acids. Thus these two experiments are used to build so called spin systems, that is build a list of resonances of the chemical shift of the peptide proton, the alpha protons and all the protons from each residue’s sidechain. Which chemical shifts corresponds to which nuclei in the spin system is determined by the conventional correlation spectroscopy connectivities and the fact that different types of protons have characteristic chemical shifts. To connect the different spinsystems in a sequential order, the nuclear Overhauser effect spectroscopy experiment has to be used. Because this experiment transfers magnetization through space, it will show crosspeaks for all protons that are close in space regardless of whether they are in the same spin system or not. The neighbouring residues are inherently close in space, so the assignments can be made by the peaks in the NOESY with other spin systems.
One important problem using homonuclear nuclear magnetic resonance is overlap between peaks. This occurs when different protons have the same or very similar chemical shifts. This problem becomes greater as the protein becomes larger, so homonuclear nuclear magnetic resonance is usually restricted to small proteins or peptides.
Nitrogen-15 nuclear magnetic resonance
The most commonly performed 15N experiment is the 1H-15N HSQC. The experiment is highly sensitive and therefore can be performed relatively quickly. It is often used to check the suitability of a protein for structure determination using NMR, as well as for the optimization of the sample conditions. It is one of the standard suite of experiments used for the determination of the solution structure of protein. The HSQC can be further expanded into three- and four dimensional NMR experiments, such as 15N-TOCSY-HSQC and 15N-NOESY-HSQC.[5]
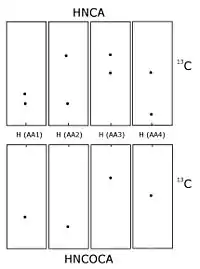
Carbon-13 and nitrogen-15 nuclear magnetic resonance
When the protein is labelled with carbon-13 and nitrogen-15 it is possible to record triple resonance experiments that transfer magnetisation over the peptide bond, and thus connect different spin systems through bonds.[6][7] This is usually done using some of the following experiments, HNCO, HN(CA)CO, HNCA,[8] HN(CO)CA, HNCACB and CBCA(CO)NH. All six experiments consist of a 1H-15N plane (similar to a HSQC spectrum) expanded with a carbon dimension. In the HN(CA)CO, each HN plane contains the peaks from the carbonyl carbon from its residue as well the preceding one in the sequence. The HNCO contains the carbonyl carbon chemical shift from only the preceding residue, but is much more sensitive than HN(CA)CO. These experiments allow each 1H-15N peak to be linked to the preceding carbonyl carbon, and sequential assignment can then be undertaken by matching the shifts of each spin system's own and previous carbons. The HNCA and HN(CO)CA works similarly, just with the alpha carbons (Cα) rather than the carbonyls, and the HNCACB and the CBCA(CO)NH contains both the alpha carbon and the beta carbon (Cβ). Usually several of these experiments are required to resolve overlap in the carbon dimension. This procedure is usually less ambiguous than the NOESY-based method since it is based on through bond transfer. In the NOESY-based methods, additional peaks corresponding to atoms that are close in space but that do not belong to sequential residues will appear, confusing the assignment process. Following the initial sequential resonance assignment, it is usually possible to extend the assignment from the Cα and Cβ to the rest of the sidechain using experiments such as HCCH-TOCSY, which is basically a TOCSY experiment resolved in an additional carbon dimension.
Restraint generation
In order to make structure calculations, a number of experimentally determined restraints have to be generated. These fall into different categories; the most widely used are distance restraints and angle restraints.
Distance restraints
A crosspeak in a NOESY experiment signifies spatial proximity between the two nuclei in question. Thus each peak can be converted into a maximum distance between the nuclei, usually between 1.8 and 6 angstroms. The intensity of a NOESY peak is proportional to the distance to the minus 6th power, so the distance is determined according to the intensity of the peak. The intensity-distance relationship is not exact, so usually a distance range is used.
It is of great importance to assign the NOESY peaks to the correct nuclei based on the chemical shifts. If this task is performed manually it is usually very labor-intensive, since proteins usually have thousands of NOESY peaks. Some computer programs such as PASD[9][10]/XPLOR-NIH,[11][12] UNIO,[13] CYANA,[14] ARIA[15]/CNS,[16] and AUDANA[17]/PONDEROSA-C/S[18] in the Integrative NMR platform[19] perform this task automatically on manually pre-processed listings of peak positions and peak volumes, coupled to a structure calculation. Direct access to the raw NOESY data without the cumbersome need of iteratively refined peak lists is so far only granted by the PASD[10] algorithm implemented in XPLOR-NIH,[11] the ATNOS/CANDID approach implemented in the UNIO software package,[13] and the PONDEROSA-C/S and thus indeed guarantees objective and efficient NOESY spectral analysis.
To obtain as accurate assignments as possible, it is a great advantage to have access to carbon-13 and nitrogen-15 NOESY experiments, since they help to resolve overlap in the proton dimension. This leads to faster and more reliable assignments, and in turn to better structures.
Angle restraints
In addition to distance restraints, restraints on the torsion angles of the chemical bonds, typically the psi and phi angles, can be generated. One approach is to use the Karplus equation, to generate angle restraints from coupling constants. Another approach uses the chemical shifts to generate angle restraints. Both methods use the fact that the geometry around the alpha carbon affects the coupling constants and chemical shifts, so given the coupling constants or the chemical shifts, a qualified guess can be made about the torsion angles.
Orientation restraints
The analyte molecules in a sample can be partially ordered with respect to the external magnetic field of the spectrometer by manipulating the sample conditions. Common techniques include addition of bacteriophages or bicelles to the sample, or preparation of the sample in a stretched polyacrylamide gel. This creates a local environment that favours certain orientations of nonspherical molecules. Normally in solution NMR the dipolar couplings between nuclei are averaged out because of the fast tumbling of the molecule. The slight overpopulation of one orientation means that a residual dipolar coupling remains to be observed. The dipolar coupling is commonly used in solid state NMR and provides information about the relative orientation of the bond vectors relative to a single global reference frame. Typically the orientation of the N-H vector is probed in an HSQC-like experiment. Initially, residual dipolar couplings were used for refinement of previously determined structures, but attempts at de novo structure determination have also been made.[20]
Hydrogen–deuterium exchange
NMR spectroscopy is nucleus specific. Thus it can distinguish between hydrogen and deuterium. The amide protons in the protein exchange readily with the solvent, and, if the solvent contains a different isotope, typically deuterium, the reaction can be monitored by NMR spectroscopy. How rapidly a given amide exchanges reflects its solvent accessibility. Thus amide exchange rates can give information on which parts of the protein are buried, hydrogen-bonded, etc. A common application is to compare the exchange of a free form versus a complex. The amides that become protected in the complex, are assumed to be in the interaction interface.
Structure calculation
The experimentially determined restraints can be used as input for the structure calculation process. Researchers, using computer programs such as XPLOR-NIH,[11] CYANA or GeNMR attempt to satisfy as many of the restraints as possible, in addition to general properties of proteins such as bond lengths and angles. The algorithms convert the restraints and the general protein properties into energy terms, and then try to minimize this energy. The process results in an ensemble of structures that, if the data were sufficient to dictate a certain fold, will converge.
Structure validation
Is important to note that the ensemble of structures obtained is an "experimental model", i.e., a representation of certain kind of experimental data. To acknowledge this fact is really important because it means that the model could be a good or bad representation of that experimental data.[21] In general, the quality of a model will depend on both the quantity and quality of experimental data used to generate it and the correct interpretation of such data.
It is important to remember that every experiment has associated errors. Random errors will affect the reproducibility and precision of the resulting structures. If the errors are systematic, the accuracy of the model will be affected. The precision indicates the degree of reproducibility of the measurement and is often expressed as the variance of the measured data set under the same conditions. The accuracy, however, indicates the degree to which a measurement approaches its "true" value.
Ideally, a model of a protein will be more accurate the more fit the actual molecule that represents and will be more precise as there is less uncertainty about the positions of their atoms. In practice there is no "standard molecule" against which to compare models of proteins, so the accuracy of a model is given by the degree of agreement between the model and a set of experimental data. Historically, the structures determined by NMR have been, in general, of lower quality than those determined by X-ray diffraction. This is due, in part, to the lower amount of information contained in data obtained by NMR. Because of this fact, it has become common practice to establish the quality of NMR ensembles, by comparing it against the unique conformation determined by X-ray diffraction, for the same protein. However, the X-ray diffraction structure may not exist, and, since the proteins in solution are flexible molecules, a protein represented by a single structure may lead to underestimate the intrinsic variation of the atomic positions of a protein. A set of conformations, determined by NMR or X-ray crystallography may be a better representation of the experimental data of a protein than a unique conformation.[22]
The utility of a model will be given, at least in part, by the degree of accuracy and precision of the model. An accurate model with relatively poor precision could be useful to study the evolutionary relationships between the structures of a set of proteins, whereas the rational drug design requires both precise and accurate models. A model that is not accurate, regardless of the degree of precision with which it was obtained will not be very useful.[21][23]
Since protein structures are experimental models that can contain errors, it is very important to be able to detect these errors. The process aimed at the detection of errors is known as validation. There are several methods to validate structures, some are statistical like PROCHECK and WHAT IF while others are based on physical principles as CheShift, or a mixture of statistical and physics principles PSVS.
Dynamics
In addition to structures, nuclear magnetic resonance can yield information on the dynamics of various parts of the protein. This usually involves measuring relaxation times such as T1 and T2 to determine order parameters, correlation times, and chemical exchange rates. NMR relaxation is a consequence of local fluctuating magnetic fields within a molecule. Local fluctuating magnetic fields are generated by molecular motions. In this way, measurements of relaxation times can provide information of motions within a molecule on the atomic level. In NMR studies of protein dynamics, the nitrogen-15 isotope is the preferred nucleus to study because its relaxation times are relatively simple to relate to molecular motions This, however, requires isotope labeling of the protein. The T1 and T2 relaxation times can be measured using various types of HSQC-based experiments. The types of motions that can be detected are motions that occur on a time-scale ranging from about 10 picoseconds to about 10 nanoseconds. In addition, slower motions, which take place on a time-scale ranging from about 10 microseconds to 100 milliseconds, can also be studied. However, since nitrogen atoms are found mainly in the backbone of a protein, the results mainly reflect the motions of the backbone, which is the most rigid part of a protein molecule. Thus, the results obtained from nitrogen-15 relaxation measurements may not be representative of the whole protein. Therefore, techniques utilising relaxation measurements of carbon-13 and deuterium have recently been developed, which enables systematic studies of motions of the amino acid side-chains in proteins. A challenging and special case of study regarding dynamics and flexibility of peptides and full-length proteins is represented by disordered structures. Nowadays, it is an accepted concept that proteins can exhibit a more flexible behaviour known as disorder or lack of structure; however, it is possible to describe an ensemble of structures instead of a static picture representing a fully functional state of the protein. Many advances are represented in this field in particular in terms of new pulse sequences, technological improvement, and rigorous training of researchers in the field.
NMR spectroscopy on large proteins
Traditionally, nuclear magnetic resonance spectroscopy has been limited to relatively small proteins or protein domains. This is in part caused by problems resolving overlapping peaks in larger proteins, but this has been alleviated by the introduction of isotope labelling and multidimensional experiments. Another more serious problem is the fact that in large proteins the magnetization relaxes faster, which means there is less time to detect the signal. This in turn causes the peaks to become broader and weaker, and eventually disappear. Two techniques have been introduced to attenuate the relaxation: transverse relaxation optimized spectroscopy (TROSY)[24] and deuteration[25] of proteins. By using these techniques it has been possible to study proteins in complex with the 900 kDa chaperone GroES-GroEL.[26]
Automation of the process
Structure determination by NMR has traditionally been a time-consuming process, requiring interactive analysis of the data by a highly trained scientist. There has been considerable interest in automating the process to increase the throughput of structure determination and to make protein NMR accessible to non-experts (See structural genomics). The two most time-consuming processes involved are the sequence-specific resonance assignment (backbone and side-chain assignment) and the NOE assignment tasks. Several different computer programs have been published that target individual parts of the overall NMR structure determination process in an automated fashion. Most progress has been achieved for the task of automated NOE assignment. So far, only the FLYA and the UNIO approach were proposed to perform the entire protein NMR structure determination process in an automated manner without any human intervention.[13][14] Recently, modules in the NMRFAM-SPARKY such as APES (two-letter-code: ae), I-PINE/PINE-SPARKY (two-letter-code: ep; I-PINE web server) and PONDEROSA (two-letter-code: c3, up; PONDEROSA web server) are integrated so that it offers full automation with visual verification capability in each step.[27] Efforts have also been made to standardize the structure calculation protocol to make it quicker and more amenable to automation.[28]
See also
References
- Wüthrich K (November 2001). "The way to NMR structures of proteins". Nature Structural & Molecular Biology. 8 (11): 923–5. doi:10.1038/nsb1101-923. PMID 11685234. S2CID 26153265.
- Clore, G. Marius (2011). "Adventures in Biomolecular NMR" (PDF). In Harris, Robin K; Wasylishen, Roderick L (eds.). Encyclopedia of Magnetic Resonance. John Wiley & Sons. doi:10.1002/9780470034590. hdl:11693/53364. ISBN 9780470034590.
- Wüthrich K (December 1990). "Protein structure determination in solution by NMR spectroscopy". J. Biol. Chem. 265 (36): 22059–62. PMID 2266107.
- Clore GM, Gronenborn AM (1989). "Determination of three-dimensional structures of proteins and nucleic acids in solution". CRC Critical Reviews in Biochemistry and Molecular Biology. 24 (5): 479–564. doi:10.3109/10409238909086962. PMID 2676353.
- Clore GM, Gronenborn AM (1991). "Structures of larger proteins in solution: three- and four-dimensional heteronuclear NMR spectroscopy". Science. 252 (5011): 1390–1399. doi:10.1126/science.2047852. OSTI 83376. PMID 2047852.
- Clore GM, Gronenborn AM (1991). "Applications of three- and four-dimensional heteronuclear NMR spectroscopy to protein structure determination". Progress in Nuclear Magnetic Resonance Spectroscopy. 23 (1): 43–92. doi:10.1016/0079-6565(91)80002-J.
- Bax A, Grzesiek S (1993). "Methodological advances in protein NMR". Accounts of Chemical Research. 26 (4): 131–138. doi:10.1021/ar00028a001.
- Bax A, Ikura M (May 1991). "An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins". J. Biomol. NMR. 1 (1): 99–104. doi:10.1007/BF01874573. PMID 1668719. S2CID 20037190.
- Kuszewski J, Schwieters CD, Garrett DS, Byrd RA, Tjandra N, Clore GM (2004). "Completely automated, highly error-tolerant macromolecular structure determination from multidimensional nuclear Overhauser enhancement spectra and chemical shift assignments". Journal of the American Chemical Society. 126 (20): 6258–6273. doi:10.1021/ja049786h. PMID 15149223.
- Kuszewski J, Thottungal RA, Clore GM, Schwieters CD (2008). "Automated error-tolerant macromolecular structure determination from multidimensional nuclear Overhauser enhancement spectra and chemical shift assignments: improved robustness and performance of the PASD algorithm". Journal of Biomolecular NMR. 41 (4): 221–239. doi:10.1007/s10858-008-9255-1. PMC 2575051. PMID 18668206.
- Schwieters CD; Kuszewski JJ; Tjandra N; Clore GM (January 2003). "The Xplor-NIH NMR molecular structure determination package". J. Magn. Reson. 160 (1): 65–73. Bibcode:2003JMagR.160...65S. doi:10.1016/S1090-7807(02)00014-9. PMID 12565051.
- Schwieters, CD; Kuszewski, JJ; Clore, GM (2006). "Using Xplor-NIH for NMR molecular structure determination". Progress in Nuclear Magnetic Resonance Spectroscopy. 48 (1): 47–62. doi:10.1016/j.pnmrs.2005.10.001.
- Herrmann T (2010). "Protein structure calculation and automated NOE restraints". Encyclopedia of Magnetic Resonance. doi:10.1002/9780470034590.emrstm1151. ISBN 978-0470034590.
- Güntert P (2004). "Automated NMR structure calculation with CYANA". Protein NMR Techniques. Methods Mol. Biol. 278. pp. 353–78. CiteSeerX 10.1.1.332.4843. doi:10.1385/1-59259-809-9:353. ISBN 978-1-59259-809-0. PMID 15318003.
- Rieping W; Habeck M; Bardiaux B; Bernard A; Malliavin TE; Nilges M (February 2007). "ARIA2: automated NOE assignment and data integration in NMR structure calculation". Bioinformatics. 23 (3): 381–2. doi:10.1093/bioinformatics/btl589. PMID 17121777.
- Brünger, AT; Adams, PD; Clore, GM; DeLano, WL; Gros, P; Grosse-Kunstleve, RW; Jiang, JS; Kuszewski, J; Nilges, M; Pannu, NS; Read, RJ; Rice, LM; Simonson, T; Warren, GL (1 September 1998). "Crystallography & NMR system: A new software suite for macromolecular structure determination". Acta Crystallographica Section D. 54 (Pt 5): 905–21. doi:10.1107/s0907444998003254. PMID 9757107.
- Lee W, Petit CM, Cornilescu G, Stark JL, Markley JL (June 2016). "The AUDANA algorithm for automated protein 3D structure determination from NMR NOE data". J. Biomol. NMR. 65 (2): 51–7. doi:10.1007/s10858-016-0036-y. ISSN 0925-2738. PMC 4921114. PMID 27169728.
- Lee, Woonghee; Stark, Jaime L.; Markley, John L. (2014-11-01). "PONDEROSA-C/S: client–server based software package for automated protein 3D structure determination". Journal of Biomolecular NMR. 60 (2–3): 73–75. doi:10.1007/s10858-014-9855-x. ISSN 0925-2738. PMC 4207954. PMID 25190042.
- Lee, Woonghee; Cornilescu, Gabriel; Dashti, Hesam; Eghbalnia, Hamid R.; Tonelli, Marco; Westler, William M.; Butcher, Samuel E.; Henzler-Wildman, Katherine A.; Markley, John L. (2016-04-01). "Integrative NMR for biomolecular research". Journal of Biomolecular NMR. 64 (4): 307–332. doi:10.1007/s10858-016-0029-x. ISSN 0925-2738. PMC 4861749. PMID 27023095.
- de Alba E; Tjandra N (2004). Residual dipolar couplings in protein structure determination. Methods Mol. Biol. 278. pp. 89–106. doi:10.1385/1-59259-809-9:089. ISBN 978-1-59259-809-0. PMID 15317993.
- Laskowski, R. A. (2003). "Structural quality assurance". Structural Bioinformatics. Methods of Biochemical Analysis. 44. pp. 273–303. doi:10.1002/0471721204.ch14. ISBN 9780471202004. PMID 12647391.
- Arnautova, Y. A.; Vila, J. A.; Martin, O. A.; Scheraga, H. A. (2009). "What can we learn by computing 13Calpha chemical shifts for X-ray protein models?". Acta Crystallographica Section D. 65 (7): 697–703. doi:10.1107/S0907444909012086. PMC 2703576. PMID 19564690.
- Spronk, C. A.; Nabuurs, S. B.; Krieger, E.; Vriend, G.; Vuister, G. W. (2004). "Validation of protein structures derived by NMR spectroscopy". Progress in Nuclear Magnetic Resonance Spectroscopy. 45 (3–4): 315–337. doi:10.1016/j.pnmrs.2004.08.003.
- Pervushin K; Riek R; Wider G; Wüthrich K (November 1997). "Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution". Proc. Natl. Acad. Sci. U.S.A. 94 (23): 12366–71. Bibcode:1997PNAS...9412366P. doi:10.1073/pnas.94.23.12366. PMC 24947. PMID 9356455.
- Markus MA; Dayie KT; Matsudaira P; Wagner G (October 1994). "Effect of deuteration on the amide proton relaxation rates in proteins. Heteronuclear NMR experiments on villin 14T". J Magn Reson B. 105 (2): 192–5. Bibcode:1994JMRB..105..192M. doi:10.1006/jmrb.1994.1122. PMID 7952934.
- Fiaux J; Bertelsen EB; Horwich AL; Wüthrich K (July 2002). "NMR analysis of a 900K GroEL GroES complex". Nature. 418 (6894): 207–11. doi:10.1038/nature00860. PMID 12110894. S2CID 2451574.
- Lee, Woonghee; Tonelli, Marco; Markley, John L. (2015-04-15). "NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy". Bioinformatics. 31 (8): 1325–1327. doi:10.1093/bioinformatics/btu830. ISSN 1367-4803. PMC 4393527. PMID 25505092.
- Liu G, Shen Y; Atreya HS; et al. (July 2005). "NMR data collection and analysis protocol for high-throughput protein structure determination". Proc. Natl. Acad. Sci. U.S.A. 102 (30): 10487–92. Bibcode:2005PNAS..10210487L. doi:10.1073/pnas.0504338102. PMC 1180791. PMID 16027363.
Further reading
- T. Kevin Hitchens; Gordon S. Rule (2005). Fundamentals of Protein NMR Spectroscopy (Focus on Structural Biology). Berlin: Springer. ISBN 978-1-4020-3499-2.
- Quincy Teng (2005). Structural biology: practical NMR applications. Berlin: Springer. Bibcode:2005stbi.book.....T. ISBN 978-0-387-24367-2.
- Mark Rance; Cavanagh, John; Wayne J. Fairbrother; Arthur W. Hunt III; Skelton, NNicholas J. (2007). Protein NMR spectroscopy: principles and practice (2nd ed.). Boston: Academic Press. ISBN 978-0-12-164491-8.
- Kurt Wüthrich (1986). NMR of proteins and nucleic acids. New York: Wiley. ISBN 978-0-471-82893-8.
External links
| Library resources about Nuclear magnetic resonance spectroscopy of proteins |
- NOESY-Based Strategy for Assignments of Backbone and Side Chain Resonances of Large Proteins without Deuteration (a protocol)
- relax Software for the analysis of NMR dynamics
- ProSA-web Archived 2011-05-11 at the Wayback Machine Web service for the recognition of errors in experimentally or theoretically determined protein structures
- Protein structure determination from sparse experimental data - an introductory presentation
- Protein NMR Protein NMR experiments