Phyre
Phyre and Phyre2 (Protein Homology/AnalogY Recognition Engine; pronounced as 'fire') are free web-based services for protein structure prediction.[1][2][3] Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times.[4] Like other remote homology recognition techniques (see protein threading), it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.[5]
| Developer(s) |
|
|---|---|
| Stable release | 2.0
/ 23 February 2011 |
| Written in | |
| Available in | English |
| Type | Bioinformatics tool for protein structure prediction |
| License | Creative Commons Attribution-2.0 |
| Website | www |
Description
The Phyre and Phyre2 servers predict the three-dimensional structure of a protein sequence using the principles and techniques of homology modeling. Because the structure of a protein is more conserved in evolution than its amino acid sequence, a protein sequence of interest (the target) can be modeled with reasonable accuracy on a very distantly related sequence of known structure (the template), provided that the relationship between target and template can be discerned through sequence alignment. Currently the most powerful and accurate methods for detecting and aligning remotely related sequences rely on profiles or hidden Markov models (HMMs). These profiles/HMMs capture the mutational propensity of each position in an amino acid sequence based on observed mutations in related sequences and can be thought of as an 'evolutionary fingerprint' of a particular protein.
Typically, the amino acid sequences of a representative set of all known three-dimensional protein structures is compiled, and these sequences are processed by scanning against a large protein sequence database. The result is a database of profiles or HMMs, one for each known 3D structure. A user sequence of interest is similarly processed to form a profile/HMM. This user profile is then scanned against the database of profiles using profile-profile or HMM-HMM alignment techniques. These alignments can also take into account patterns of predicted or known secondary structure elements and can be scored using various statistical models. See protein structure prediction for more information.
The first Phyre server was released in June 2005 and uses a profile-profile alignment algorithm based on each protein's position-specific scoring matrix.[6] The Phyre2 server was publicly released February 2011 as a replacement for the original Phyre server and provides extra functionality over Phyre, a more advanced interface, fully updated fold library and uses the HHpred / HHsearch package for homology detection among other improvements.
Standard usage
After pasting a protein amino acid sequence into the Phyre or Phyre2 submission form, a user will typically wait between 30 minutes and several hours (depending on factors such as sequence length, number of homologous sequences and frequency and length of insertions and deletions) for a prediction to complete. An email containing summary information and the predicted structure in PDB format are sent to the user together with a link to a web page of results. The Phyre2 results screen is divided into three main sections, described below.
Secondary structure and disorder prediction
The user-submitted protein sequence is first scanned against a large sequence database using PSI-BLAST. The profile generated by PSI-BLAST is then processed by the neural network secondary structure prediction program PsiPred[7] and the protein disorder predictor Disopred.[8] The predicted presence of alpha-helices, beta-strands and disordered regions is shown graphically together with a color-coded confidence bar.
Domain analysis

Many proteins contain multiple protein domains. Phyre2 provides a table of template matches color-coded by confidence and indicating the region of the user sequence matched. This can aid in the determination of the domain composition of a protein.
Detailed template information
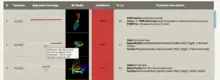
The main results table in Phyre2 provides confidence estimates, images and links to the three-dimensional predicted models and information derived from either Structural Classification of Proteins database (SCOP) or the Protein Data Bank (PDB) depending on the source of the detected template. For each match a link takes the user to a detailed view of the alignment between the user sequence and the sequence of known three-dimensional structure.
Alignment view

The detailed alignment view permits a user to examine individual aligned residues, matches between predicted and known secondary structure elements and the ability to toggle information regarding patterns of sequence conservation and secondary structure confidence. In addition Jmol is used to permit interactive 3D viewing of the protein model.
Improvements in Phyre2
Phyre2 uses a fold library that is updated weekly as new structures are solved. It uses a more up-to-date interface and offers additional functionality over the Phyre server as described below.
Batch processing
The batch processing feature permits users to submit more than one sequence to Phyre2 by uploading a file of sequences in FASTA format. By default, users have a limit of 100 sequences in a batch. This limit can be raised by contacting the administrator. Batch jobs are processed in the background on free computing power as it becomes available. Thus, batch jobs will often take longer than individually submitted jobs, but this is necessary to allow a fair distribution of computing resources to all Phyre2 users.
One to one threading
One to one threading allows you to upload both a sequence you wish modelled AND the template on which to model it. Users sometimes have a protein sequence that they wish to model on a specific template of their choice. This may be for example a newly solved structure that is not in the Phyre2 database or because of some additional biological information that indicates the chosen template would produce a more accurate model than the one(s) automatically chosen by Phyre2.
Backphyre
Instead of predicting the 3D structure of a protein sequence, often users have a solved structure and they are interested in determining if there is a related structure in a genome of interest. In Phyre2 an uploaded protein structure can be converted into a hidden Markov model and then scanned against a set of genomes (more than 20 genomes as of March 2011). This functionality is called "BackPhyre" to indicate how Phyre2 is being used in reverse.
Phyrealarm
Sometimes Phyre2 can't detect any confident matches to known structures. However, the fold library database increases by about 40-100 new structures each week. So even though there might be no decent templates this week, there may well be in the coming weeks. Phyrealarm allows users to submit a protein sequence to be automatically scanned against new entries added to the fold library every week. If a confident hit is detected, the user is automatically notified by email together with the results of the Phyre2 search. Users can also control the level of alignment coverage and confidence in the match required to trigger an email alert.
3DLigandSite
Phyre2 is coupled to the 3DLigandSite[9] server for protein binding site prediction. 3DLigandSite has been one of the top performing servers for binding site prediction at the Critical Assessment of Techniques for Protein Structure Prediction (CASP) in (CASP8 and CASP9). Confident models produced by Phyre2 (confidence >90%) are automatically submitted to 3DLigandSite.
Transmembrane topology prediction
The program memsat_svm[10] is used to predict the presence and topology of any transmembrane helices present in the user protein sequence.
Multi-template modelling
Phyre2 permits users to choose 'Intensive' modelling from the main submission screen. This mode:
- Examines the list of hits and applies heuristics in order to select templates that maximise sequence coverage and confidence.
- Constructs models for each selected template.
- Uses these models to provide pairwise distance constraints that are input to the ab initio and multi-template modelling tool Poing.[11]
- Poing synthesises the user protein in the context of these distance constraints, modelled by springs. Regions for which there is no template information are modelled by the ab initio simplified physics model of Poing.
- The complete model generated by Poing is combined with the original templates as input to MODELLER.
Applications
Applications of Phyre and Phyre2 include protein structure prediction, function prediction, domain prediction, domain boundary prediction, evolutionary classification of proteins, guiding site-directed mutagenesis and solving protein crystal structures by molecular replacement.
There are two linked resources that use Phyre predictions for the structure-based analysis of missense variants typically resulting from single-nucleotide polymorphisms.
- PhyreRisk is a database which maps genetic variants to experimental and Phyre-predicted protein structures. The protein page displays the experimental and predicted structures. Users can map variants from either genetic or protein coordinates.[12]
- Missense3D is a tool which provides a stereochemical report on the effect of a missense variant on protein structure. Users can upload their own variants and coordinates, including both PDB structures and Phyre-predicted models.[13]
History
Phyre and Phyre2 are the successors to the 3D-PSSM[14] protein structure prediction system which has over 1400 citations to date.[15] 3D-PSSM was designed and developed by Lawrence Kelley[16] and Bob MacCallum[17] in the Biomolecular modelling Lab[18] at the Cancer Research UK. Phyre and Phyre2 were Lawrence Kelley in the Structural bioinformatics group,[19] Imperial College London. Components of the Phyre and Phyre2 systems were developed by Benjamin Jefferys,[20] Alex Herbert,[21] and Riccardo Bennett-Lovsey.[22] Research and development of both servers was supervised by Michael Sternberg.
References
- Lawrence Kelley; Riccardo Bennett-Lovsey; Alex Herbert; Kieran Fleming. "Phyre: Protein Homology/analogY Recognition Engine". Structural Bioinformatics Group, Imperial College, London. Retrieved 22 April 2011.
- Lawrence Kelley; Benjamin Jefferys. "Phyre2: Protein Homology/analogY Recognition Engine V 2.0". Structural Bioinformatics Group, Imperial College, London. Retrieved 22 April 2011.
- Kelley, L. A.; Sternberg, M. J. E. (2009). "Protein structure prediction on the Web: A case study using the Phyre server" (PDF). Nature Protocols. 4 (3): 363–71. doi:10.1038/nprot.2009.2. hdl:10044/1/18157. PMID 19247286. S2CID 12497300.
- Number of results returned from a search on Google Scholar (Google Scholar search)
- "Help: About PHYRE2". PHYRE Protein Fold Recognition Server.
The work on developing the Phyre2 web server is supported by a BBSRC tools and resources grant
- Bennett-Lovsey, R. M.; Herbert, A. D.; Sternberg, M. J. E.; Kelley, L. A. (2007). "Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre". Proteins: Structure, Function, and Bioinformatics. 70 (3): 611–25. doi:10.1002/prot.21688. PMID 17876813. S2CID 23530683.
- McGuffin, L. J.; Bryson, K.; Jones, D. T. (2000). "The PSIPRED protein structure prediction server". Bioinformatics. 16 (4): 404–5. doi:10.1093/bioinformatics/16.4.404. PMID 10869041.
- Jones, D. T.; Ward, J. J. (2003). "Prediction of disordered regions in proteins from position specific score matrices". Proteins: Structure, Function, and Genetics. 53: 573–8. doi:10.1002/prot.10528. PMID 14579348. S2CID 6081008.
- Wass, M. N.; Kelley, L. A.; Sternberg, M. J. E. (2010). "3DLigand Site: Predicting ligand-binding sites using similar structures". Nucleic Acids Research. 38 (Web Server issue): W469–W473. doi:10.1093/nar/gkq406. PMC 2896164. PMID 20513649.
- Jones, D. T. (2007). "Improving the accuracy of transmembrane protein topology prediction using evolutionary information". Bioinformatics. 23 (5): 538–44. doi:10.1093/bioinformatics/btl677. PMID 17237066.
- Jefferys, B. R.; Kelley, L. A.; Sternberg, M. J. E. (2010). "Protein Folding Requires Crowd Control in a Simulated Cell". Journal of Molecular Biology. 397 (5): 1329–38. doi:10.1016/j.jmb.2010.01.074. PMC 2891488. PMID 20149797.
- Ofoegbu, Tochukwu C.; David, Alessia; Kelley, Lawrence A.; Mezulis, Stefans; Islam, Suhail A.; Mersmann, Sophia F.; Strömich, Léonie; Vakser, Ilya A.; Houlston, Richard S.; Sternberg, Michael J.E. (2019). "PhyreRisk: A Dynamic Web Application to Bridge Genomics, Proteomics and 3D Structural Data to Guide Interpretation of Human Genetic Variants". Journal of Molecular Biology. 431 (13): 2460–2466. doi:10.1016/j.jmb.2019.04.043. ISSN 0022-2836. PMC 6597944. PMID 31075275.
- Ittisoponpisan, Sirawit; Islam, Suhail A.; Khanna, Tarun; Alhuzimi, Eman; David, Alessia; Sternberg, Michael J.E. (2019). "Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated?". Journal of Molecular Biology. 431 (11): 2197–2212. doi:10.1016/j.jmb.2019.04.009. ISSN 0022-2836. PMC 6544567. PMID 30995449.
- Kelley, L. A.; MacCallum, R. M.; Sternberg, M. J. E. (2000). "Enhanced genome annotation using structural profiles in the program 3D-PSSM". Journal of Molecular Biology. 299 (2): 501–522. doi:10.1006/jmbi.2000.3741. PMID 10860755.
- Number of results returned from a search on Google Scholar. (Google Scholar search)
- Dr. Lawrence Kelley
- Dr. Bob Maccallum
- "Biomolecular Modelling laboratory". Archived from the original on 2011-09-29. Retrieved 2011-03-09.
- Structural Bioinformatics Group
- "Dr. Benjamin Jefferys". Archived from the original on 2011-04-18. Retrieved 2011-03-28.
- Dr. Alex Herbert
- Dr. Riccardo Bennett-Lovsey