Protein function prediction
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.[1]
Generally, function can be thought of as, "anything that happens to or through a protein".[1] The Gene Ontology Consortium provides a useful classification of functions, based on a dictionary of well-defined terms divided into three main categories of molecular function, biological process and cellular component.[2] Researchers can query this database with a protein name or accession number to retrieve associated Gene Ontology (GO) terms or annotations based on computational or experimental evidence.
While techniques such as microarray analysis, RNA interference, and the yeast two-hybrid system can be used to experimentally demonstrate the function of a protein, advances in sequencing technologies have made the rate at which proteins can be experimentally characterized much slower than the rate at which new sequences become available.[3] Thus, the annotation of new sequences is mostly by prediction through computational methods, as these types of annotation can often be done quickly and for many genes or proteins at once. The first such methods inferred function based on homologous proteins with known functions (homology-based function prediction). The development of context-based and structure based methods have expanded what information can be predicted, and a combination of methods can now be used to get a picture of complete cellular pathways based on sequence data.[3] The importance and prevalence of computational prediction of gene function is underlined by an analysis of 'evidence codes' used by the GO database: as of 2010, 98% of annotations were listed under the code IEA (inferred from electronic annotation) while only 0.6% were based on experimental evidence.[4]
Function prediction methods
Homology-based methods

Proteins of similar sequence are usually homologous[5] and thus have a similar function. Hence proteins in a newly sequenced genome are routinely annotated using the sequences of similar proteins in related genomes.
However, closely related proteins do not always share the same function.[6] For example, the yeast Gal1 and Gal3 proteins are paralogs (73% identity and 92% similarity) that have evolved very different functions with Gal1 being a galactokinase and Gal3 being a transcriptional inducer.[7]
There is no hard sequence-similarity threshold for "safe" function prediction; many proteins of barely detectable sequence similarity have the same function while others (such as Gal1 and Gal3) are highly similar but have evolved different functions. As a rule of thumb, sequences that are more than 30-40% identical are usually considered as having the same or a very similar function.
For enzymes, predictions of specific functions are especially difficult, as they only need a few key residues in their active site, hence very different sequences can have very similar activities. By contrast, even with sequence identity of 70% or greater, 10% of any pair of enzymes have different substrates; and differences in the actual enzymatic reactions are not uncommon near 50% sequence identity.[8][9]
Sequence motif-based methods
The development of protein domain databases such as Pfam (Protein Families Database)[10] allow us to find known domains within a query sequence, providing evidence for likely functions. The dcGO website[11] contains annotations to both the individual domains and supra-domains (i.e., combinations of two or more successive domains), thus via dcGO Predictor allowing for the function predictions in a more realistic manner. Within protein domains, shorter signatures known as 'motifs' are associated with particular functions,[12] and motif databases such as PROSITE ('database of protein domains, families and functional sites') can be searched using a query sequence.[13] Motifs can, for example, be used to predict subcellular localization of a protein (where in the cell the protein is sent after synthesis). Short signal peptides direct certain proteins to a particular location such as the mitochondria, and various tools exist for the prediction of these signals in a protein sequence.[14] For example, SignalP, which has been updated several times as methods are improved.[15] Thus, aspects of a protein's function can be predicted without comparison to other full-length homologous protein sequences.
Structure-based methods
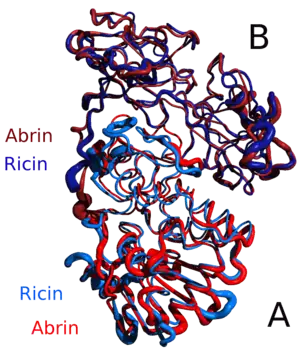
Because 3D protein structure is generally more well conserved than protein sequence, structural similarity is a good indicator of similar function in two or more proteins.[6][12] Many programs have been developed to screen an unknown protein structure against the Protein Data Bank[16] and report similar structures (for example, FATCAT (Flexible structure AlignmenT by Chaining AFPs (Aligned Fragment Pairs) with Twists),[17] CE (combinatorial extension)[18]) and DeepAlign (protein structure alignment beyond spatial proximity).[19] To deal with the situation that many protein sequences have no solved structures, some function prediction servers such as RaptorX are also developed that can first predict the 3D model of a sequence and then use structure-based method to predict functions based upon the predicted 3D model. In many cases instead of the whole protein structure, the 3D structure of a particular motif representing an active site or binding site can be targeted.[12][20][21][22][23] The Structurally Aligned Local Sites of Activity (SALSA) [21] method, developed by Mary Jo Ondrechen and students, utilizes computed chemical properties of the individual amino acids to identify local biochemically active sites. Databases such as Catalytic Site Atlas[24] have been developed that can be searched using novel protein sequences to predict specific functional sites.
Genomic context-based methods
Many of the newer methods for protein function prediction are not based on comparison of sequence or structure as above, but on some type of correlation between novel genes/proteins and those that already have annotations. Also known as phylogenomic profiling, these genomic context based methods are based on the observation that two or more proteins with the same pattern of presence or absence in many different genomes most likely have a functional link.[12][25] Whereas homology-based methods can often be used to identify molecular functions of a protein, context-based approaches can be used to predict cellular function, or the biological process in which a protein acts.[3][25] For example, proteins involved in the same signal transduction pathway are likely to share a genomic context across all species.
Gene fusion
Gene fusion occurs when two or more genes encode two or more proteins in one organism and have, through evolution, combined to become a single gene in another organism (or vice versa for gene fission).[3][26] This concept has been used, for example, to search all E. coli protein sequences for homology in other genomes and find over 6000 pairs of sequences with shared homology to single proteins in another genome, indicating potential interaction between each of the pairs.[26] Because the two sequences in each protein pair are non-homologous, these interactions could not be predicted using homology-based methods.
Co-location/co-expression
In prokaryotes, clusters of genes that are physically close together in the genome often conserve together through evolution, and tend to encode proteins that interact or are part of the same operon.[3] Thus, chromosomal proximity also called the gene neighbour method[27] can be used to predict functional similarity between proteins, at least in prokaryotes. Chromosomal proximity has also been seen to apply for some pathways in selected eukaryotic genomes, including Homo sapiens,[28] and with further development gene neighbor methods may be valuable for studying protein interactions in eukaryotes.[25]
Genes involved in similar functions are also often co-transcribed, so that an unannotated protein can often be predicted to have a related function to proteins with which it co-expresses.[12] The guilt by association algorithms developed based on this approach can be used to analyze large amounts of sequence data and identify genes with expression patterns similar to those of known genes.[29][30] Often, a guilt by association study compares a group of candidate genes (unknown function) to a target group (for example, a group of genes known to be associated with a particular disease), and rank the candidate genes by their likelihood of belonging to the target group based on the data.[31] Based on recent studies, however, it has been suggested that some problems exist with this type of analysis. For example, because many proteins are multifunctional, the genes encoding them may belong to several target groups. It is argued that such genes are more likely to be identified in guilt by association studies, and thus predictions are not specific.[31]
With the accumulation of RNA-seq data that are capable of estimating expression profiles for alternatively spliced isoforms, machine learning algorithms have also been developed for predicting and differentiating functions at the isoform level.[32] This represents an emerging research area in function prediction, which integrates large-scale, heterogeneous genomic data to infer functions at the isoform level.[33]
Computational solvent mapping
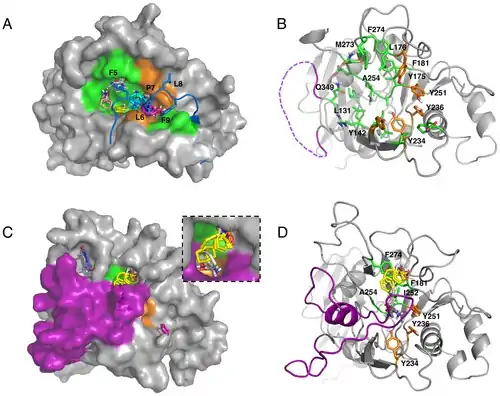
One of the challenges involved in protein function prediction is discovery of the active site. This is complicated by certain active sites not being formed – essentially existing – until the protein undergoes conformational changes brought on by the binding of small molecules. Most protein structures have been determined by X-ray crystallography which requires a purified protein crystal. As a result, existing structural models are generally of a purified protein and as such lack the conformational changes that are created when the protein interacts with small molecules.[35]
Computational solvent mapping utilizes probes (small organic molecules) that are computationally 'moved' over the surface of the protein searching for sites where they tend to cluster. Multiple different probes are generally applied with the goal being to obtain a large number of different protein-probe conformations. The generated clusters are then ranked based on the cluster's average free energy. After computationally mapping multiple probes, the site of the protein where relatively large numbers of clusters form typically corresponds to an active site on the protein.[35]
This technique is a computational adaptation of 'wet lab' work from 1996. It was discovered that ascertaining the structure of a protein while it is suspended in different solvents and then superimposing those structures on one another produces data where the organic solvent molecules (that the proteins were suspended in) typically cluster at the protein's active site. This work was carried out as a response to realizing that water molecules are visible in the electron density maps produced by X-ray crystallography. The water molecules are interacting with the protein and tend to cluster at the protein's polar regions. This led to the idea of immersing the purified protein crystal in other solvents (e.g. ethanol, isopropanol, etc.) to determine where these molecules cluster on the protein. The solvents can be chosen based on what they approximate, that is, what molecule this protein may interact with (e.g. ethanol can probe for interactions with the amino acid serine, isopropanol a probe for threonine, etc.). It is vital that the protein crystal maintains its tertiary structure in each solvent. This process is repeated for multiple solvents and then this data can be used to try to determine potential active sites on the protein.[36] Ten years later this technique was developed into an algorithm by Clodfelter et al.
Network-based methods
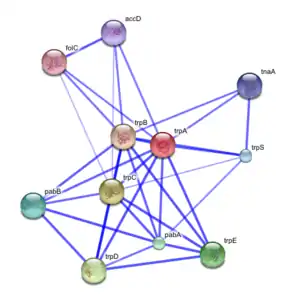
Guilt by association type algorithms may be used to produce a functional association network for a given target group of genes or proteins. These networks serve as a representation of the evidence for shared/similar function within a group of genes, where nodes represent genes/proteins and are linked to each other by edges representing evidence of shared function.[37]
Integrated networks
Several networks based on different data sources can be combined into a composite network, which can then be used by a prediction algorithm to annotate candidate genes or proteins.[38] For example, the developers of the bioPIXIE system used a wide variety of Saccharomyces cerevisiae (yeast) genomic data to produce a composite functional network for that species.[39] This resource allows the visualization of known networks representing biological processes, as well as the prediction of novel components of those networks. Many algorithms have been developed to predict function based on the integration of several data sources (e.g. genomic, proteomic, protein interaction, etc.), and testing on previously annotated genes indicates a high level of accuracy.[37][40] Disadvantages of some function prediction algorithms have included a lack of accessibility, and the time required for analysis. Faster, more accurate algorithms such as GeneMANIA (multiple association network integration algorithm) have however been developed in recent years[38] and are publicly available on the web, indicating the future direction of function prediction.
Tools and databases for protein function prediction
STRING: web tool that integrates various data sources for function prediction.[41]
VisANT: Visual analysis of networks and integrative visual data-mining.[42]
References
- Rost B, Liu J, Nair R, Wrzeszczynski KO, Ofran Y (December 2003). "Automatic prediction of protein function". Cellular and Molecular Life Sciences. 60 (12): 2637–50. doi:10.1007/s00018-003-3114-8. PMID 14685688. S2CID 8800506.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (May 2000). "Gene ontology: tool for the unification of biology. The Gene Ontology Consortium". Nature Genetics. 25 (1): 25–9. doi:10.1038/75556. PMC 3037419. PMID 10802651.
- Gabaldón T, Huynen MA (April 2004). "Prediction of protein function and pathways in the genome era". Cellular and Molecular Life Sciences. 61 (7–8): 930–44. doi:10.1007/s00018-003-3387-y. PMID 15095013. S2CID 18032660.
- du Plessis L, Skunca N, Dessimoz C (November 2011). "The what, where, how and why of gene ontology--a primer for bioinformaticians". Briefings in Bioinformatics. 12 (6): 723–35. doi:10.1093/bib/bbr002. PMC 3220872. PMID 21330331.
- Reeck GR, de Haën C, Teller DC, Doolittle RF, Fitch WM, Dickerson RE, et al. (August 1987). ""Homology" in proteins and nucleic acids: a terminology muddle and a way out of it". Cell. 50 (5): 667. doi:10.1016/0092-8674(87)90322-9. PMID 3621342. S2CID 42949514.
- Whisstock JC, Lesk AM (August 2003). "Prediction of protein function from protein sequence and structure". Quarterly Reviews of Biophysics. 36 (3): 307–40. doi:10.1017/S0033583503003901. PMID 15029827.
- Platt A, Ross HC, Hankin S, Reece RJ (March 2000). "The insertion of two amino acids into a transcriptional inducer converts it into a galactokinase". Proceedings of the National Academy of Sciences of the United States of America. 97 (7): 3154–9. Bibcode:2000PNAS...97.3154P. doi:10.1073/pnas.97.7.3154. PMC 16208. PMID 10737789.
- Rost B (April 2002). "Enzyme function less conserved than anticipated". Journal of Molecular Biology. 318 (2): 595–608. doi:10.1016/S0022-2836(02)00016-5. PMID 12051862.
- Tian W, Skolnick J (October 2003). "How well is enzyme function conserved as a function of pairwise sequence identity?". Journal of Molecular Biology. 333 (4): 863–82. CiteSeerX 10.1.1.332.4052. doi:10.1016/j.jmb.2003.08.057. PMID 14568541.
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A (January 2010). "The Pfam protein families database". Nucleic Acids Research. 38 (Database issue): D211–22. doi:10.1093/nar/gkp985. PMC 2808889. PMID 19920124.
- Fang H, Gough J (January 2013). "DcGO: database of domain-centric ontologies on functions, phenotypes, diseases and more". Nucleic Acids Research. 41 (Database issue): D536–44. doi:10.1093/nar/gks1080. PMC 3531119. PMID 23161684.
- Sleator RD, Walsh P (March 2010). "An overview of in silico protein function prediction". Archives of Microbiology. 192 (3): 151–5. doi:10.1007/s00203-010-0549-9. PMID 20127480. S2CID 8932206.
- Sigrist CJ, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N (January 2010). "PROSITE, a protein domain database for functional characterization and annotation". Nucleic Acids Research. 38 (Database issue): D161–6. doi:10.1093/nar/gkp885. PMC 2808866. PMID 19858104.
- Menne KM, Hermjakob H, Apweiler R (August 2000). "A comparison of signal sequence prediction methods using a test set of signal peptides". Bioinformatics. 16 (8): 741–2. doi:10.1093/bioinformatics/16.8.741. PMID 11099261.
- Petersen TN, Brunak S, von Heijne G, Nielsen H (September 2011). "SignalP 4.0: discriminating signal peptides from transmembrane regions". Nature Methods. 8 (10): 785–6. doi:10.1038/nmeth.1701. PMID 21959131. S2CID 16509924.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (January 2000). "The Protein Data Bank". Nucleic Acids Research. 28 (1): 235–42. doi:10.1093/nar/28.1.235. PMC 102472. PMID 10592235.
- Ye Y, Godzik A (July 2004). "FATCAT: a web server for flexible structure comparison and structure similarity searching". Nucleic Acids Research. 32 (Web Server issue): W582–5. doi:10.1093/nar/gkh430. PMC 441568. PMID 15215455.
- Shindyalov IN, Bourne PE (September 1998). "Protein structure alignment by incremental combinatorial extension (CE) of the optimal path". Protein Engineering. 11 (9): 739–47. doi:10.1093/protein/11.9.739. PMID 9796821.
- Wang S, Ma J, Peng J, Xu J (March 2013). "Protein structure alignment beyond spatial proximity". Scientific Reports. 3: 1448. Bibcode:2013NatSR...3E1448W. doi:10.1038/srep01448. PMC 3596798. PMID 23486213.
- Parasuram R, Lee JS, Yin P, Somarowthu S, Ondrechen MJ (December 2010). "Functional classification of protein 3D structures from predicted local interaction sites". Journal of Bioinformatics and Computational Biology. 8 Suppl 1: 1–15. doi:10.1142/s0219720010005166. PMID 21155016.
- Wang Z, Yin P, Lee JS, Parasuram R, Somarowthu S, Ondrechen MJ (2013). "Protein function annotation with Structurally Aligned Local Sites of Activity (SALSAs)". BMC Bioinformatics. 14 Suppl 3: S13. doi:10.1186/1471-2105-14-S3-S13. PMC 3584854. PMID 23514271.
- Garma LD, Juffer AH (April 2016). "Comparison of non-sequential sets of protein residues". Computational Biology and Chemistry. 61: 23–38. doi:10.1016/j.compbiolchem.2015.12.004. PMID 26773655.
- Garma LD, Medina M, Juffer AH (November 2016). "Structure-based classification of FAD binding sites: A comparative study of structural alignment tools". Proteins. 84 (11): 1728–1747. doi:10.1002/prot.25158. PMID 27580869. S2CID 26066208.
- Porter CT, Bartlett GJ, Thornton JM (January 2004). "The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data". Nucleic Acids Research. 32 (Database issue): D129–33. doi:10.1093/nar/gkh028. PMC 308762. PMID 14681376.
- Eisenberg D, Marcotte EM, Xenarios I, Yeates TO (June 2000). "Protein function in the post-genomic era". Nature. 405 (6788): 823–6. doi:10.1038/35015694. PMID 10866208. S2CID 4398864.
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D (July 1999). "Detecting protein function and protein-protein interactions from genome sequences". Science. 285 (5428): 751–3. CiteSeerX 10.1.1.535.9650. doi:10.1126/science.285.5428.751. PMID 10427000.
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N (March 1999). "The use of gene clusters to infer functional coupling". Proceedings of the National Academy of Sciences of the United States of America. 96 (6): 2896–901. Bibcode:1999PNAS...96.2896O. doi:10.1073/pnas.96.6.2896. PMC 15866. PMID 10077608.
- Lee JM, Sonnhammer EL (May 2003). "Genomic gene clustering analysis of pathways in eukaryotes". Genome Research. 13 (5): 875–82. doi:10.1101/gr.737703. PMC 430880. PMID 12695325.
- Walker MG, Volkmuth W, Sprinzak E, Hodgson D, Klingler T (December 1999). "Prediction of gene function by genome-scale expression analysis: prostate cancer-associated genes". Genome Research. 9 (12): 1198–203. doi:10.1101/gr.9.12.1198. PMC 310991. PMID 10613842.
- Klomp JA, Furge KA (July 2012). "Genome-wide matching of genes to cellular roles using guilt-by-association models derived from single sample analysis". BMC Research Notes. 5 (1): 370. doi:10.1186/1756-0500-5-370. PMC 3599284. PMID 22824328.
- Pavlidis P, Gillis J (2012). "Progress and challenges in the computational prediction of gene function using networks". F1000Research. 1 (14): 14. doi:10.3410/f1000research.1-14.v1. PMC 3782350. PMID 23936626.
- Eksi R, Li HD, Menon R, Wen Y, Omenn GS, Kretzler M, Guan Y (Nov 2013). "Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data". PLOS Computational Biology. 9 (11): e1003314. Bibcode:2013PLSCB...9E3314E. doi:10.1371/journal.pcbi.1003314. PMC 3820534. PMID 24244129.
- Li HD, Menon R, Omenn GS, Guan Y (August 2014). "The emerging era of genomic data integration for analyzing splice isoform function". Trends in Genetics. 30 (8): 340–7. doi:10.1016/j.tig.2014.05.005. PMC 4112133. PMID 24951248.
- Wang G, MacRaild CA, Mohanty B, Mobli M, Cowieson NP, Anders RF, Simpson JS, McGowan S, Norton RS, Scanlon MJ (2014). "Molecular insights into the interaction between Plasmodium falciparum apical membrane antigen 1 and an invasion-inhibitory peptide". PLOS ONE. 9 (10): e109674. Bibcode:2014PLoSO...9j9674W. doi:10.1371/journal.pone.0109674. PMC 4208761. PMID 25343578.
- Clodfelter KH, Waxman DJ, Vajda S (August 2006). "Computational solvent mapping reveals the importance of local conformational changes for broad substrate specificity in mammalian cytochromes P450". Biochemistry. 45 (31): 9393–407. doi:10.1021/bi060343v. PMID 16878974.
- Mattos C, Ringe D (May 1996). "Locating and characterizing binding sites on proteins". Nature Biotechnology. 14 (5): 595–9. doi:10.1038/nbt0596-595. PMID 9630949. S2CID 20273975.
- Sharan R, Ulitsky I, Shamir R (2007). "Network-based prediction of protein function". Molecular Systems Biology. 3 (88): 88. doi:10.1038/msb4100129. PMC 1847944. PMID 17353930.
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q (2008). "GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function". Genome Biology. 9 Suppl 1: S4. doi:10.1186/gb-2008-9-s1-s4. PMC 2447538. PMID 18613948.
- Myers CL, Robson D, Wible A, Hibbs MA, Chiriac C, Theesfeld CL, Dolinski K, Troyanskaya OG (2005). "Discovery of biological networks from diverse functional genomic data". Genome Biology. 6 (13): R114. doi:10.1186/gb-2005-6-13-r114. PMC 1414113. PMID 16420673.
- Peña-Castillo L, Tasan M, Myers CL, Lee H, Joshi T, Zhang C, Guan Y, Leone M, Pagnani A, Kim WK, Krumpelman C, Tian W, Obozinski G, Qi Y, Mostafavi S, Lin GN, Berriz GF, Gibbons FD, Lanckriet G, Qiu J, Grant C, Barutcuoglu Z, Hill DP, Warde-Farley D, Grouios C, Ray D, Blake JA, Deng M, Jordan MI, Noble WS, Morris Q, Klein-Seetharaman J, Bar-Joseph Z, Chen T, Sun F, Troyanskaya OG, Marcotte EM, Xu D, Hughes TR, Roth FP (2008). "A critical assessment of Mus musculus gene function prediction using integrated genomic evidence". Genome Biology. 9 Suppl 1 (S1): S2. doi:10.1186/gb-2008-9-s1-s2. PMC 2447536. PMID 18613946.
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C (January 2017). "The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible". Nucleic Acids Research. 45 (D1): D362–D368. doi:10.1093/nar/gkw937. PMC 5210637. PMID 27924014.
- Granger BR, Chang YC, Wang Y, DeLisi C, Segrè D, Hu Z (April 2016). "Visualization of Metabolic Interaction Networks in Microbial Communities Using VisANT 5.0". PLOS Computational Biology. 12 (4): e1004875. Bibcode:2016PLSCB..12E4875G. doi:10.1371/journal.pcbi.1004875. PMC 4833320. PMID 27081850.
External links
- The dcGO database
- Protein Data Bank
- Catalytic Site Atlas
- RaptorX Server for model-assisted protein function prediction
- Blast2GO, high-throughput tool for protein function prediction and functional annotation (webpage).