Supramolecular catalysis
Supramolecular catalysis is not a well-defined field but it generally refers to an application of supramolecular chemistry, especially molecular recognition and guest binding, toward catalysis.[1][2] This field was originally inspired by enzymatic system which, unlike classical organic chemistry reactions, utilizes non-covalent interactions such as hydrogen bonding, cation-pi interaction, and hydrophobic forces to dramatically accelerate rate of reaction and/or allow highly selective reactions to occur. Because enzymes are structurally complex and difficult to modify, supramolecular catalysts offer a simpler model for studying factors involved in catalytic efficiency of the enzyme.[3]:1 Another goal that motivates this field is the development of efficient and practical catalysts that may or may not have an enzyme equivalent in nature.

A closely related field of study is asymmetric catalysis which requires molecular recognition to differentiate two chiral starting material or chiral transition states and thus it could be categorized as an area of supramolecular catalysis, but supramolecular catalysis however does not necessarily have to involve asymmetric reaction. As there is another Wikipedia article already written about small molecule asymmetric catalysts, this article focuses primarily on large catalytic host molecules. Non-discrete and structurally poorly defined system such as micelle and dendrimers are not included.
History
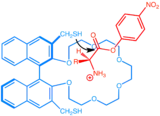
The term supramolecular chemistry is defined by Jean-Marie Lehn as "the chemistry of intermolecular bond, covering structures and functions of the entities formed by association of two or more chemical species" in his Nobel lecture in 1987,[6] but the concept of supramolecular catalysis was started way earlier in 1946 by Linus Pauling when he founded the theory of enzymatic catalysis in which rate acceleration is the result of non-covalent stabilization of the transition state by the enzymes.[7] Nevertheless, it was not until a few decades later that an artificial enzyme was developed. The first simple enzyme mimics were based on crown ether and cryptand.[8] In 1976, less than ten years after the discovery of crown ether, Cram et al. developed a functionalized binapthyl crown ether that catalyze transacylation.[4] The catalyst makes use the crown ether motif's ability to capture cation to bind to the ammonium ion part of the substrate and subsequently employs the nearby thiol motif to cleave the ester.
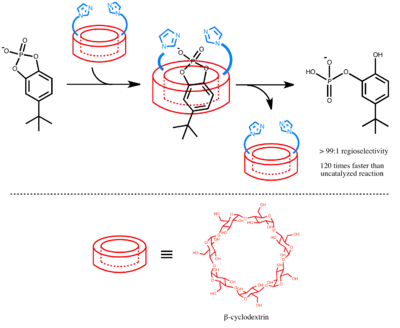
From the early 1970s, cyclodextrins have been extensively studied for its encapsulation properties and used as binding sites in supramolecular catalyst.[2] Cyclodextrins have rigid ring structure, hydrophilic surface, and hydrophobic cavity on the inside; therefore, they are capable of binding organic molecules in aqueous solution. In 1978, with the background knowledge that the hydrolysis of m-tert-butylphenyl acetate is accelerated in the presence of 2-benzimidazoleacetic acid and alpha-cyclodextrin,[9] Brewslow et al. developed a catalyst based on a beta-cyclodextrin carrying two imidazole groups. This cyclodextrin catalytic system mimics ribonuclease A by its use of a neutral imidazole and an imidazolium cation to selective cleave cyclic phosphate substrates. The rate of the reaction is catalyzed 120 times faster, and unlike a hydrolysis by simple base NaOH that gives a 1:1 mixture of the products, this catalysts yield a 99:1 selectivity for one compound.[5]
In 1993, Rebek et al. developed the first self-assemble capsule[10] and in 1997 the so-called "tennis ball" structure was used to catalyze a Diels-Alder reaction.[11] Self-assembled molecules have an advantage over crown ether and cyclodextrin in that they can capture significant larger molecules or even two molecules at the same time. In the following decades, many research groups, such as Makoto Fujita, Ken Raymond, and Jonathan Nitschke, developed cage-like catalysts also from molecular self-assembly principle.
In 2002, Sanders and coworkers published the use of dynamic combinatorial library technique to construct a receptor[12] and in 2003 they employed the technique to develop a catalyst for Diels-Alder reaction.[13]
Mechanism of catalysis
Three common modes of catalysis are described here.
Orienting reactive and labile groups
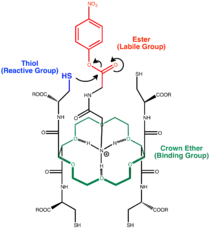
A supramolecular host could bind to a guest molecule in such a way that the guest's labile group is positioned close to the reactive group of the host. The proximity of the two groups enhances the probability that the reaction could occur and thus the reaction rate is increased. This concept is similar to the principle of preorganization which states that complexation could be improved if the binding motifs are preorganized in a well-defined position so that the host does not require any major conformational change for complexation.[15] In this case, the catalyst is preorganized such that no major conformational changes is required for the reaction to occur. A notable example of catalysts that employ this mechanism is Jean-Marie Lehn's crown ether.[14] In addition, catalysts based on functionalized cyclodextrins often employ this mode of catalysis.[16]:88
Raising the effective substrate concentration
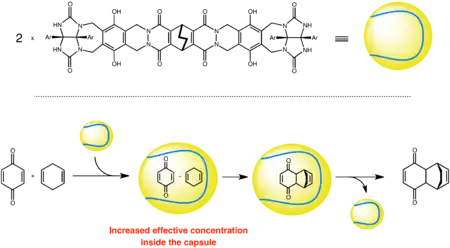
Bimolecular reactions are highly dependent on the concentration of substrates. Therefore, when a supramolecular container encapsulates both reactants within its small cavity, the effective local concentration of the reactants is increased and, as a result of an entropic effect, the rate of the reaction is accelerated.[16]:89 That is to say an intramolecular reaction is faster than its corresponding intermolecular reaction.
Although high raise in effective concentration is observed, molecules that employ this mode of catalysis have tiny rate acceleration compared to that of enzymes. A proposed explanation is that in a container the substrates are not as tightly bound as in enzyme. The reagents have room to wiggle in a cavity and so the entropic effect might not be as important. Even in the case of enzymes, computational studies have shown that the entropic effect might also be overestimated.[17]
Examples of molecules that work via this mechanism are Rebek's tennis ball and Fujita's octahedral complex.[11][18]
Stabilizing transition state
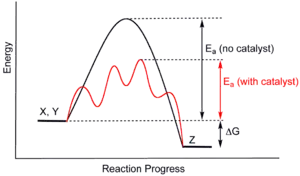
Supramolecular catalysts can accelerate reactions not only by placing the two reactants in close proximity but also by stabilizing the transition state of the reaction and reducing activation energy.[16]:89 While this fundamental principle of catalysis is common in small molecule or heterogeneous catalysts, supramolecular catalysts however has a difficult time utilizing the concept due to their often rigid structures. Unlike enzymes that can change shape to accommodate the substrates, supramolecules do not have that kind of flexibility and so rarely achieve sub-angstrom adjustment required for perfect transition state stabilization.[3]:2
An example of catalysts of this type is Sander's porphyrin trimer. A Diels Alder reaction between two pyridine functionalized substrates normally yield a mixture of endo and exo products. In the presence of the two catalysts, however, complete endo selectivity or exo selectivity could be obtained. The underlying cause of the selectivity is the coordination interaction between pyridine and the zinc ion on porphyrin. Depending on the shape of the catalysts, one product is preferred over the other.[19]
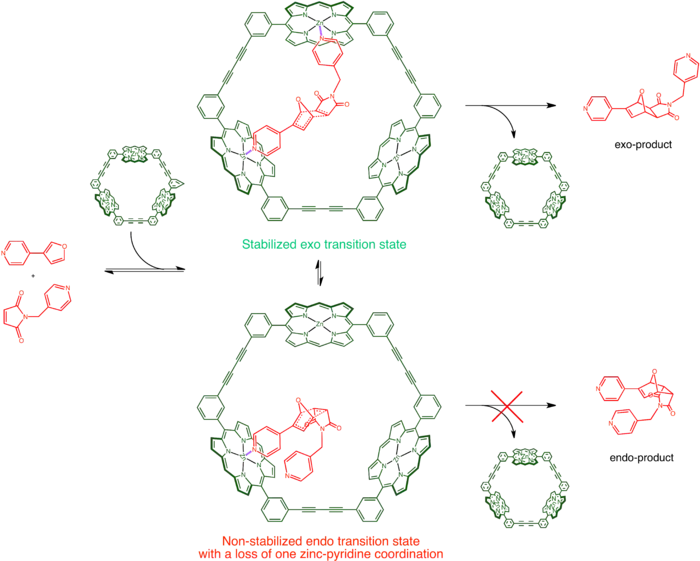
Approaches to making supramolecular catalysts
Design approach
The traditional approach to supramolecular catalysts focuses on the design of macromolecular receptor with appropriately placed catalytic functional groups. These catalysts are often inspired by the structure of enzymes with the catalytic group mimicking reactive amino acid residues, but unlike real enzymes, the binding sites of these catalysts are rigid structure made from chemical building blocks.[20] All of the examples in this article are developed via the design approach.
Jeremy Sanders pointed out that the design approach has not been successful and has produced very few efficient catalysts because of rigidity of the supramolecules. He argued that rigid molecules with a slight mismatch to the transition state cannot be an efficient catalyst. Rather than investing so much synthesis effort on one rigid molecule that we cannot determine its precise geometry to the sub-angstrom level which is required for good stabilization, Sanders suggested the use of many small flexible building blocks with competing weak interactions so that it is possible for the catalyst to adjust its structure to accommodate the substrate better.[21] There is a direct trade-off between the enthalpic benefit from flexible structure and the entropic benefit from rigid structure.[3]:3 Flexible structure could perhaps bind the transition state better but it allows more room for the substrates to move and vibrate. Most supramolecular chemists in the past prefer to build rigid structures out of fear of entropic cost.[21]
This problem could perhaps be mended by Baker and Houk's "inside-out approach" which allows a systematic de novo enzyme development.[22] This computational method starts simply with a predicted transition state structure and slowly builds outward by optimizing the arrangement of functional groups to stabilize the transition state. Then it fills out the remainder of the active site and, finally, it generates an entire protein scaffold that could contain the designed active site. This method could potentially be applied to supramolecular catalysis, although a plethora of chemical building blocks could easily overwhelm the computational model intended to work with 20 amino acids.
Transition state analogue selection/screening approach
Assuming that catalytic activity largely depends on the catalyst's affinity to the transition state, one could synthesize a transition state analog (TSA), a structure that resembles the transition state of the reaction. Then one could link the TSA to a solid-support or identifiable tag and use that TSA to select an optimal catalyst from a mixture of many different potential catalysts generated chemically or biologically by a diversity oriented synthesis. This method allows quick screening of a library of diverse compounds. It does not require as much synthetic effort and it allows a study of various catalytic factors simultaneously. Hence the method could potentially yield an efficient catalyst that we could not have designed with our current knowledge.[20]
Many catalytic antibodies were developed and studied using this approach.
Catalytic activity screening approach

A problem with transition state analogue selection approach is that catalytic activity is not a screening criterion. TSAs do not necessarily represent real transition states and so a catalyst obtained from screening could just be the best receptor for a TSA but is not necessarily the best catalyst. To circumvent this problem, catalytic activity needs to be measured directly and also quickly. To develop a high-throughput screen, substrates could be designed to change color or release a fluorescent product upon reaction. For example, Crabtree and coworkers utilized this method in screening for a hydrosylation catalysts for alkene and imine.[23] Unfortunately the prerequisite for such substrates narrow down the range of reactions for study.[20]
Dynamic combinatorial library approach
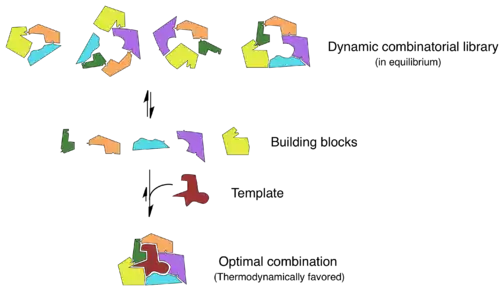
In contrast to traditional combinatorial synthesis where a library of catalysts were first generated and later screened (as in the two above approaches), dynamic combinatorial library approach utilizes a mixture of multicomponent building blocks that reversibly form library of catalysts. With out a template, the library consists of a roughly equal mixture of different combination of building blocks. In the presence of a template which is either a starting material or a TSA, the combination that provides the best binding to the template is thermodynamically favorable and thus that combination is more prevalent than other library members. The biased ratio of the desired catalyst to other combinatorial products could then be frozen by terminating the reversibility of the equilibrium by means such as change in temperature, pH, or radiation to yield the optimal catalyst.[20] For example, Lehn et al. used this method to create a dynamic combinatorial library of imine inhibitor from a set of amines and a set of aldehydes. After some time, the equilibrium was terminated by an addition of NaBH3CN to afford the desired catalyst.[24]
Prominent examples of supramolecular catalysts
Diederich's pyruvate oxidase mimic
In nature, pyruvate oxidase employs two cofactors thiamine pyrophosphate (ThDP) and Flavin adenine dinucleotide (FAD) to catalyze a conversion of pyruvate to acetyl phosphate. First, ThDP mediates a decarboxylation of pyruvate and generates an active aldehyde as a product. The aldehyde is then oxidized by FAD and is subsequently attacked by phosphate to yield acetyl phosphate.
Diederich and coworkers mimicked this system with a supramolecular catalyst based on cyclophane. The catalyst has thiazolium ion, a reactive part of ThDP and flavin, a bare-bones core of FAD, in close proximity and near the substrate binding site. The catalytic cycle is almost the same as that in nature, except the substrate is an aromatic aldehyde rather than pyruvate. First, the catalyst binds the substrate within its cyclophane ring. Then, it uses thiazolium ion to condense with the substrate generating an active aldehyde. This aldehyde is oxidized by flavin and then attacked by methanol to yield a methyl ester.[25]
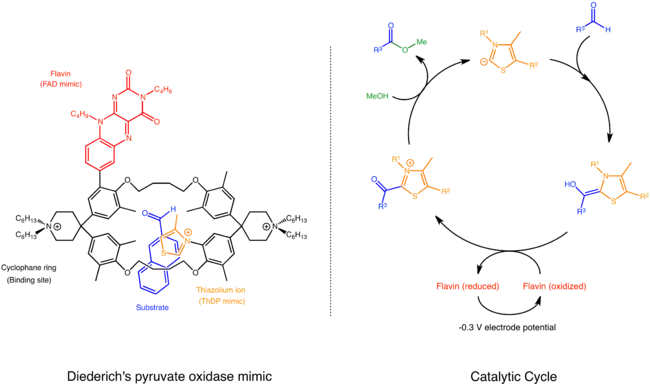
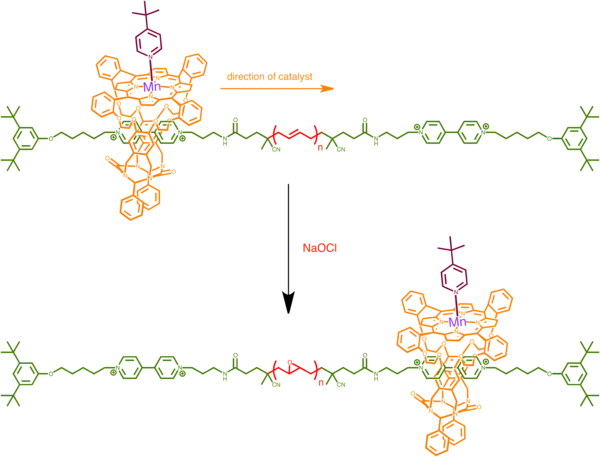
Nolte's successive epoxidation catalyst for alkene polymer
Processive enzymes are proteins that catalyze consecutive reactions without releasing its substrate. An example of processive enzymes is RNA polymerase which binds to a DNA strand and repeatedly catalyzes nucleotide transfers, effectively synthesizing a corresponding RNA strand.
Nolte and coworkers developed an artificial processive enzyme in a form of manganese porphyrin rotaxane that treads along a long polymer of alkene and catalyze multiple rounds of alkene epoxidation. Manganese (III) ion in the porphyrin is the molecule's catalytic center, capable of epoxidation in the presence of an oxygen donor and an activating ligand. With a small ligand such pyridine that binds manganese from inside the cavity of the rotaxane, epoxidation happens outside the catalyst. With a large bulky ligand such as tert-butyl pyridine that does not fit inside the cavity however, epoxidation happens on the inside of the catalyst.[26]
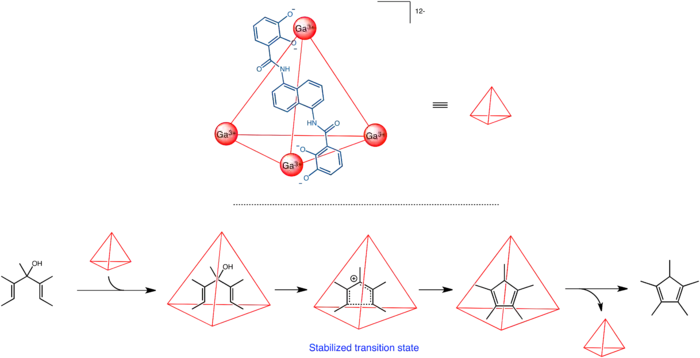
Raymond's Nazarov cyclization catalyst
Raymond and coworkers developed a supramolecular host M4L6 (4 gallium ions and 6 ligands for each complex) that self-assembles via metal-ligand interaction in aqueous solution. This container molecule is polyanionic and thus its tetrahedron-shaped cavity is capable of encapsulating and stabilizing a cationic molecule. Consequently, encapsulated molecule can be easily protonated as a resulting carbocation from protonation is stabilized by the surrounding anions. Raymond utilized this property to perform acid-catalyzed Nazarov cyclization. The catalyst accelerates the reaction by over one million fold, making it the most efficient supramolecular catalyst to date. It was proposed that such a high catalytic activity does not arise just from the increased basicity of the encapsulated substrate but also from the constrictive binding that stabilize the transition state of the cyclization. Unfortunately, this catalyst has a problem with product inhibition. To by pass that problem, the product of the cyclization reaction could be reacted with a dienophile transforming it into a Diels-Alder adduct that no longer fits inside the catalyst cavity.[1]
In this case, the supramolecular host was initially designed to simply capture cationic guests. Almost a decade later, it was exploited as a catalyst for Nazarov cyclization.
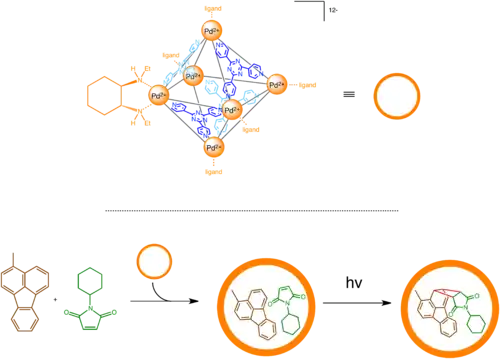
Fujita's chiral self-assembled catalyst for asymmetric [2+2] photoadditions
Fujita and coworkers discovered a self-assemble M6L4 (6 palladium ions and 4 ligands in each complex) supramolecular container that could be enhanced into a chiral supramolecule by an addition of peripheral chiral auxiliary. In this case, the auxiliary diethyldiaminocyclohexane does not directly activate the catalytic site but induces a slight deformation of the triazine plane to create chiral cavity inside the container molecule. This container could then be used to asymmetrically catalyze a [2+2] photoaddition of maleimide and inert aromatic compound fluoranthene, which previously have not been shown to undergo thermal or photochemical pericyclic reaction. The catalyst yields an enantiomeric excess of 40%.[27]
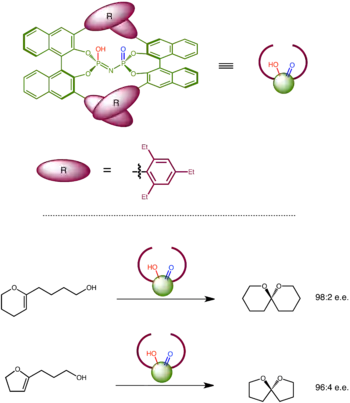
List's confined Bronsted acid as a catalyst for asymmetric spiroacetalization
Inspired by enzymes with deep active site pocket, List and coworkers designed and constructed a set of confined Bronsted acids with an extremely sterically demanding chiral pocket based on a C2-symmetric bis(binapthyl) imidodiphosphoric acid. Within the chiral microenvironment, the catalysts has a geometrically fixed bifunctional active site that activates both an electrophilic part and a nucleophilic part of a substrate. This catalyst enables stereoselective spiroacetal formation with high enantiomeric excess for a variety of substrates.[28]
Supramolecular inhibitors
Supramolecular containers do not only have an application in catalysis but also in the opposite, namely, inhibition. A container molecule could encapsulate a guest molecule and thus subsequently renders the guest unreactive. A mechanism of inhibition could either be that the substrate is completely isolated from the reagent or that the container molecule destabilize the transition state of the reaction.
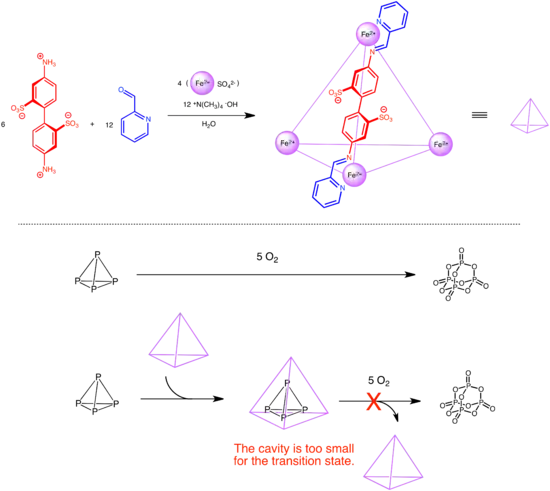
Nitschke and coworkers invented a self-assembly M4L6 supramolecular host with a tetrahedral hydrophobic cavity that can encapsulate white phosphorus. Pyrophoric phosphorus, which could self-combust upon contact with air, is rendered air-stable within the cavity. Even though the hole in the cavity is large enough for an oxygen molecule to enter, the transition state of the combustion is too large to fit within the small cage cavity.[29]
Problems and limitations
After many decades since its inception, supramolecular chemistry's application in practical catalysis remains elusive. Supramolecular catalysis has not yet made significant contribution in the area of industrial chemistry or synthetic methodology.[21] Here are few problems associated with this field.
Product inhibition
In many supramolecular catalytic systems designed to work with bimolecular addition reactions like the Diels-Alder, the product of the reaction binds more strongly to the supramolecular host than the two substrates do, consequently leading to inhibition by the product. As a result, these catalysts has a turnover number of one and are not truly catalytic. A stoichiometric quantity of the catalysts is needed for a full conversion.[30]
Poor transition state stabilization
Most supramolecular catalysts are developed from rigid building blocks because rigid blocks are less complicated than flexible parts in constructing a desired shape and placing functional groups where the designer wants. Due to the rigidity, however, a slight mismatch from the transition state inevitably leads to poor stabilization and thus poor catalysis. In nature, enzymes are flexible and could change their structures to bind a transition state better than their native form.[21]
Difficulty in synthesis and further adjustment
Syntheses of large complex catalysts are time and resource consuming. An unexpected deviation from the design could be disastrous. Once a catalyst is discovered, modification for further adjustment could be so synthetically challenging that it is easier to study the poor catalyst than to improve it.[21]
See also
References
- Raymond, K. N.; Hastings, C. J.; Pluth, M. D.; Bergman, R. G. (2010). "Enzymelike Catalysis of the Nazarov Cyclization by Supramolecular Encapsulation". Journal of the American Chemical Society. 132 (20): 6938–6940. doi:10.1021/ja102633e. PMID 20443566.
- Nolte, R. J. M.; Vriezema, D. M.; Aragone, M. C.; Elemans, J. J. A. W.; Cornelissen, J. J. L. M.; Rowan, A. E. (2005). "Self-Assembled Nanoreactors". Chemical Reviews. 105 (4): 1445–1489. doi:10.1021/cr0300688. hdl:2066/32981. PMID 15826017.
- van Leeuwen, P. W. N. M. (2008). Supramolecular Catalysis. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA. ISBN 978-3-527-32191-9.
- Cram, D. J.; Chao, Y. (1976). "Enzyme Mechanisms, Models, and Mimics". Journal of the American Chemical Society. 98 (4): 1015–1017. doi:10.1021/ja00420a026.
- Breslow, R.; Doherty, J. B.; Guillot, G.; Lipsey, C. (1978). "The Use of Cycloamylose to Probe the Charge-Relay System". Journal of the American Chemical Society. 100 (10): 3227–3229. doi:10.1021/ja00478a052.
- Lehn, J. (1988). "Selection and Amplification of a Catalyst from a Dynamic Combinatorial Library". Angewandte Chemie International Edition. 27 (1): 89–112. doi:10.1002/anie.198800891.
- Pauling, L. (1946). "Molecular Architecture and Biological Reactions" (PDF). Chemical and Engineering News. 24 (10): 1375–1377. doi:10.1021/cen-v024n010.p1375.
- Kirby, A. J. (1996). "Enzyme Mechanisms, Models, and Mimics". Angewandte Chemie International Edition. 35 (7): 706–724. doi:10.1002/anie.199607061.
- Bender, M. L.; Komiyama, M.; Breaux, E. J. (1977). "The Use of Cycloamylose to Probe the Charge-Relay System". Bioorganic Chemistry. 6 (2): 127–136. doi:10.1016/0045-2068(77)90015-3.
- Rebek, J. Jr.; Wyler, R.; de Mendoza J. (1993). "A Synthetic Cavity Assembles Through Self-Complementary Hydrogen Bonds". Angewandte Chemie International Edition. 32 (12): 1699–1701. doi:10.1002/anie.199316991.
- Rebek, J. Jr; Kang, J. (1997). "Acceleration of a Diels–Alder Reaction by a Self-Assembled Molecular Capsule". Nature. 385 (661): 50–52. Bibcode:1997Natur.385...50K. doi:10.1038/385050a0. PMID 8985245.
- Sanders, J. K. M.; Otto, S.; Furlan, R. L. E. (2002). "Selection and Amplification of Hosts From Dynamic Combinatorial Libraries of Macrocyclic Disulfides". Science. 297 (5581): 590–593. Bibcode:2002Sci...297..590O. doi:10.1126/science.1072361. PMID 12142534.
- Otto, S.; Brisig, B.; Sanders, J. K. M. (2003). "Selection and Amplification of a Catalyst from a Dynamic Combinatorial Library". Angewandte Chemie International Edition. 42 (11): 1270–1273. doi:10.1002/anie.200390326. PMID 12645061.
- Lehn, J.; Sirlin, C. (1978). "Molecular Catalysis: Enhanced Rates of Thiolysis with High Structural and Chiral Recognition in Complexes of a Reactive Macrocyclic Receptor Molecule". Chemical Communications (21): 949–951. doi:10.1039/C39780000949.
- Cram, D. J. (1988). "The Design of Molecular Hosts, Guests, and Their Complexes". Angewandte Chemie International Edition. 27 (8): 1009–1020. doi:10.1002/anie.198810093.
- Beer, P.; Gale, P. A.; Smith, D. K. (1999). Supramolecular Chemistry. New York: Oxford University Press. ISBN 978-0-19-850447-4.
- Warshel, A.; Aaqvist, J. (1993). "The Design of Molecular Hosts, Guests, and Their Complexes". Chemical Reviews. 93 (7): 2523–2544. doi:10.1021/cr00023a010.
- Fujita, M.; Yoshizawa, M.; Tamura, M. (2006). "Diels-Alder in Aqueous Molecular Hosts: Unusual Regioselectivity and Efficient Catalysis". Science. 312 (5771): 251–254. Bibcode:2006Sci...312..251Y. doi:10.1126/science.1124985. PMID 16614218.
- Sanders, J. K. M.; Walter, C. J.; Anderson, H. L. (1993). "Exo-Selective Scceleration of an Intermolecular Diels–Alder Reaction by a Trimeric Porphyrin Host". Chemical Communications (5): 458–460. doi:10.1039/C39930000458.
- Motherwell, W. B.; Bingham, M. J.; Six, Y. (2001). "Recent Progress in the Design and Synthesis of Artificial Enzymes". Tetrahedron. 57 (22): 4663–4686. doi:10.1016/S0040-4020(01)00288-5.
- Sanders, J. K. M. (1998). "Supramolecular Catalysis in Transition". Chemistry: A European Journal. 4 (8): 1378–1383. doi:10.1002/(SICI)1521-3765(19980807)4:8<1378::AID-CHEM1378>3.0.CO;2-3.
- Houk, K. N.; Kiss, G.; Çelebi-Ölçüm, N.; Moretti, R.; Baker, D. (2013). "Computational Enzyme Design". Angewandte Chemie International Edition. 52 (22): 5700–5725. doi:10.1002/anie.201204077.
- Crabtree, R. H.; Cooper, A. C.; McAlexander, L. H.; Lee, D.-H.; Torres, M. T. (1998). "Reactive Dyes as a Method for Rapid Screening of Homogeneous Catalysts". Journal of the American Chemical Society. 120 (38): 9971–9972. doi:10.1021/ja9818607.
- Lehn, J.; Huc, I. (1997). "Virtual Combinatorial Libraries: Dynamic Generation of Molecular and Supramolecular Diversity by Self-Assembly". PNAS. 94 (6): 2106–2110. Bibcode:1997PNAS...94.2106H. doi:10.1073/pnas.94.6.2106. PMC 20048. PMID 9122156.
- Diederich, F.; Mattei, P. (1997). "Catalytic Cyclophanes. Part XI. A Flavo-thiazolio-cyclophane as a Biomimetic Catalyst for the Preparative-scale Electro-oxidation of Aromatic Aldehydes to Methyl Esters". Helvetica Chimica Acta. 80 (5): 1555–1588. doi:10.1002/hlca.19970800516.
- Nolte, R. J. M.; Thordarson, P.; Bijsterveld, E. J. A.; Rowan, A. E. (2003). "Epoxidation of Polybutadiene by a Topologically Linked Catalyst". Nature. 424 (6951): 915–918. Bibcode:2003Natur.424..915T. doi:10.1038/nature01925. PMID 12931181.
- Fujita, M.; Nishioka, Y.; Yamaguchi, T.; Kawano, M. (2008). "Asymmetric [2 + 2] Olefin Cross Photoaddition in a Self-Assembled Host with Remote Chiral Auxiliaries". Journal of the American Chemical Society. 130 (26): 8160–8161. doi:10.1021/ja802818t. PMID 18540605.
- List, B.; Čorić, I. (2012). "Asymmetric Spiroacetalization Catalysed by Confined Brønsted Acids". Nature. 483 (7389): 315–319. Bibcode:2012Natur.483..315C. doi:10.1038/nature10932. PMID 22422266.
- Nitschke, J. R.; Mal, P.; Breiner, B.; Rissanen, K. (2009). "White Phosphorus Is Air-Stable Within a Self-Assembled Tetrahedral Capsule". Science. 324 (5935): 1697–1699. Bibcode:2009Sci...324.1697M. doi:10.1126/science.1175313. PMID 19556504.
- Easton, C. J.; Lincoln, S. F.; Barr, L.; Onagi H. (2004). "Molecular Reactors and Machines: Applications, Potential, and Limitations". Chemistry: A European Journal. 10 (13): 3120–3128. doi:10.1002/chem.200305768. PMID 15224320.