Thiol-ene reaction
The thiol-ene reaction (also alkene hydrothiolation) is an organic reaction between a thiol and an alkene to form a thioether. This reaction was first reported in 1905,[1] but it gained prominence in the late 1990s and early 2000s for its feasibility and wide range of applications.[2][3] This reaction is accepted as a click chemistry reaction given the reactions’ high yield, stereoselectivity, high rate, and thermodynamic driving force.

The reaction results in an anti-Markovnikov addition of a thiol compound to an alkene. Given the stereoselectivity, high rate and yields, this synthetically useful reaction may underpin future applications in material and biomedical sciences.[2][4]
Mechanisms
Radical addition
Thiol-ene additions are known to proceed through two mechanisms: free-radical additions and catalyzed Michael additions. Free-radical additions can be initiated by light, heat or radical initiators, which form a thiyl radical species. The radical then propagates with an ene functional group via an anti-Markovnikov addition to form a carbon-centered radical. A chain-transfer step removes a hydrogen radical from a thiol, which can subsequently participate in multiple propagation steps.[4]
Thiol-ene radical additions are advantageous for chemical synthesis because the step growth (propagation and chain-transfer steps) and chain growth (homopolymerization) processes can be effectively used to form homogeneous polymer networks. Photopolymerization is a useful radical-based reaction for applications within the nanotechnology, biomaterial, and material sciences, but these reactions are hindered by the inhibitory capabilities of oxygen. The thiol-ene radical addition combines the benefits of photopolymerization reactions with the aforementioned advantages of click chemistry reactions. This reaction is useful to the field of radical-based photopolymerization because it quantitatively and rapidly proceeds through a simple mechanism under ambient atmospheric conditions.[4] The carbon-centered radical can undergo chain-growth polymerization depending on the thiol and ene functional groups. This free-radical polymerization can be useful in the synthesis of uniform polymer networks.[5]
Michael addition
Thiol-ene reactions are known to proceed through a Michael addition pathway. These reactions are catalyzed by either a base or a nucleophile, resulting in a similar anti-Markovnikov addition product as the thiol-ene radical addition.[6]
Kinetics
Click chemistry reactions are known to be high efficiency and have fast reaction rates, yet there is considerable variability in the overall reaction rate depending on the functionality of the alkene. To better understand the kinetics of thiol-ene reactions, calculations and experiments of transition-state and reaction enthalpies were conducted for a number of alkenes and their radical intermediates.[5][7] It was shown that the reactivity and structure of the alkene determines whether the reaction will follow a step-growth or chain-growth pathway.[5] It was also shown that the thiol-ene polymerization can be tuned by enhancing intermolecular interactions between the thiol and alkene functional groups.[7] A currently accepted trend is that electron-rich alkenes (such as vinyl ether or allyl ether) and norbornene are highly reactive compared to conjugated and electron-poor alkenes (butadiene and methoxyethene). In the case of norbornene and vinyl ether only step-growth is observed, no homopolymerization occurs after the formation of the carbon centered radical.[4]

Due to the complex kinetics of this two-step cyclic reaction, the rate-determining step was difficult to delineate. Given that the rates of both steps must be equal, the concentration of the radical species is determined by the rate constant of the slower of the reaction steps. Thus the overall reaction rate (RP) can be modeled by the ratio of the propagation rate (kP) to the chain-transfer rate (kCT).The behavior of the reaction rate is outlined by the relationship below. In all cases the reaction is first order, when kP ≫ kCT [Eq. 1] the reaction rate is determined by the thiol concentration and the rate limiting step is chain-transfer, when kP ≪ kCT [Eq. 2] the reaction rate is determined by the alkene concentration and the rate limiting step is the propagation, and finally when kP ≈ kCT [Eq. 3] the reaction is half order with respect to both the alkene and thiol concentrations.
The functional groups on the thiol and alkene compounds can affect the reactivity of the radical species and their respective rate constants. The structure of the alkene determines whether the reaction will be propagation or chain-transfer limited, and therefore first order with respect to alkene or thiol concentration respectively. In the case of reactive alkenes, such as allyl ether, chain-transfer is the rate-limiting step, while in the case of less reactive alkenes, such as vinyl silazanes, propagation is the rate-limiting step. The thiol’s hydrogen affinity also affects the rate-limiting step. Alkyl thiols have less abstractable protons and therefore the chain-transfer step has a lower reaction rate than the propagation step.[4]
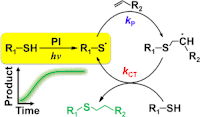
Most time the quasi-first-order reaction yields a kinetic rate equation following the exponential decay function for the reactants and products. However, when the radical generation becomes the rate-limiting step, an induction period is often observed at the early stage of the reaction, for example, for photoinitiated reaction under weak light condition. The kinetic curve deviates from the exponential decay function for a common first-order reaction by having a slow growth period. The kinetic model has to include the radical generation step to explain this induction period (right figure). The final expression has a Gaussian-like shape.[8]
- [normalized thiol-ene product] =

where k is an effective rate constant and t is time.
Synthetically useful thiol-ene reactions
Initiation of cascade cyclization
The thiol-ene reaction (and analogous thiol-yne reaction) have extensively been used in generating reactive intermediates for the cyclization of unsaturated substrates. Radical hydrothiolation of an unsaturated functional group indirectly generates a carbon-centered radical, which can then cyclize intramolecularly onto alkenes, oxime ethers, isocyanides, cyano groups, and aromatic rings.[9] The use of thiyl radicals as initiators of cyclization has been employed in the synthesis of a number of natural products, including aplysins,[10] α-kainic acid,[11] asperparalines,[12] and alkaloids such as narciclasine and lycoricidine.[13]

Intramolecular thiol-ene reactions
Intramolecular thiol-ene reactions provide a means to create sulphur-containing heterocycles. The radical-initiated thiol-ene reaction has enabled the successful synthesis of four- to eight-membered rings, as well as macrocycles. While the radical thiol-ene reaction favors the anti-Markovnikov product, the regiochemistry of the cycloaddition depends on substituent effects and reaction conditions, which serve to direct the cyclization towards the thermodynamically or kinetically favored product respectively. This section examines intramolecular thiol-ene cyclization reactions, which yields a mixture of 5-exo and 6-endo products in order to facilitate a discussion of the factors, which may affect the regioselectivity of the intramolecular addition. This reaction has relevance for the synthesis of C-linked thiosugars.[14] Both the furanose and pyranose thiosugars can be prepared from the same thiyl radical precursor; 5-exo and 6-endo cyclizations of this precursor form the respective desired compound. The conditions under which these cyclization reactions occur follow Baldwin's rules for ring closure.

Cis–trans conversion of alkenes
Given the reversibility of the thiol-ene radical addition, the reaction can be used to facilitate cis–trans isomerizations. The thiyl radical propagates with the alkene to form a carbon-centered radical, the previous double bond now allows free rotation around the single sigma bond. When the reverse reaction occurs, the orientation of the hydrogen addition on the carbon radical determines whether the isomerization product will be cis or trans. Therefore, composition of the products depends on the conformational stability of the carbon-centered radical intermediate.[15]
Potential applications
Dendrimer synthesis
Dendrimers are promising in medicine, biomaterials, and nanoengineering. These polymers can functions as targeting components, detecting agents, and pharmaceutically-active compounds. Thiol-ene additions are useful in the divergent synthesis of dendrimers due to the characteristics of click chemistry such as the mild reaction conditions (benign solvents), regioselectivity, high efficiency, high conversion and quantitative yield.[16] Because this reaction is photo-initiated, it does not require copper catalysis, unlike other common reactions used in dendrimer preparation; this is advantageous for the synthesis of functional biomaterials given the inhibitory characteristic of copper on biological systems.[17] Thiol-ene reactions have been used alongside anhydride, esterification, Grignard, and Michael reactions to functionalize chain ends and build polymer backbones in the synthesis of branched molecules such as glycodendrons, polythioether dendrimers and organosilicon thioether dendrimers.[3][4][18]
A general strategy for the divergent synthesis of a dendrimer begins with a core; commonly-used cores include 2,4,6-triallyloxy-1,3,5-triazine, triallyl isocyanurate and tetravinylsilane.[16][17][19] In a well cited report, 2,4,6-triallyloxy-1,3,5-triazine was mixed with 1-thioglycerol in the absence of solvent, the thiol-ene reaction was initiated by the radical initiator 2,2-dimethoxy-2-phenylacetophenone and UV irradiation. Terminal alkene functional groups were added to the dendrimer via esterification by pent-4-enoic anhydride in the presence of DMAP and pyridine. The fourth generation product prepared in a stepwise fashion contains 48 terminal hydroxyl groups[16]
Surface patterning
The thiol-ene functionalization of surface has been widely investigated in material science and biotechnology. The attachment of a molecule with a sterically accessible alkene or thiol group to a solid surface enables the construction of polymers on the surface through subsequent thiol-ene reactions.[2] Given that in aqueous solutions thiol-ene reactions can be initiated by UV light (wavelength 365–405 nm) or sunlight, the attachment of a given functional group to the exposed thiol or alkene can be controlled spatially through photomasking.[20] More specifically, a photomask, enables the selective exposure of a surface to a UV light source, controlling the location of a given thiol-ene reaction, whereas the identity of the attached molecule is determined by the composition of the aqueous phase placed above the surface at the time of UV exposure. Thus, the manipulation of the shape of the photomask and the composition of the aqueous layer results in the creation of heterogeneous surface, whose properties depend on identity of the attached molecule at a given location.[2]
Thiol-ene functionalization of a surface can be achieved with a high level of spatial specificity, allowing the production of photomasks.[20]
Organo-triethoxysilane molecules, either thiol or vinyl tailed, have been introduced in surface functionalization. Ethoxysilane and methoxysilane functional groups are commonly used to anchor organic molecules on a variety of oxides surfaces. The thiol-ene coupling can be achieved either in the bulk solution before molecular anchoring [8] or step-wise onto a substrate that enables photolithography.[21] The reaction can be done in five minutes under sunlight that has ~4% UV light that is useful for the thiol-ene reaction.[8]
Protein patterning on electron beam resist
Thiol-ene can also be used as an electron beam resist, resulting in nanostructures that allow direct protein functionalization.[22]
See also
References
- Posner, Theodor (January 1905). "Beiträge zur Kenntniss der ungesättigten Verbindungen. II. Ueber die Addition von Mercaptanen an ungesättigte Kohlenwasserstoffe". Berichte der Deutschen Chemischen Gesellschaft. 38 (1): 646–657. doi:10.1002/cber.190503801106.
- Lowe, Andrew B. (2010). "Thiol-ene "click" reactions and recent applications in polymer and materials synthesis". Polym. Chem. 1 (1): 17–36. doi:10.1039/B9PY00216B.
- Nilsson, Camilla; Simpson, Neil; Malkoch, Michael; Johansson, Mats; Malmström, Eva (15 February 2008). "Synthesis and thiol-ene photopolymerization of allyl-ether functionalized dendrimers". Journal of Polymer Science Part A: Polymer Chemistry. 46 (4): 1339–1348. Bibcode:2008JPoSA..46.1339N. doi:10.1002/pola.22474.
- Hoyle, Charles E.; Bowman, Christopher N. (22 February 2010). "Thiol-Ene Click Chemistry". Angewandte Chemie International Edition. 49 (9): 1540–1573. doi:10.1002/anie.200903924. PMID 20166107.
- Northrop, Brian H.; Coffey, Roderick N. (22 August 2012). "Thiol–Ene Click Chemistry: Computational and Kinetic Analysis of the Influence of Alkene Functionality". Journal of the American Chemical Society. 134 (33): 13804–13817. doi:10.1021/ja305441d. PMID 22853003.
- Nair, Devatha P.; Podgórski, Maciej; Chatani, Shunsuke; Gong, Tao; Xi, Weixian; Fenoli, Christopher R.; Bowman, Christopher N. (14 January 2014). "The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry". Chemistry of Materials. 26 (1): 724–744. doi:10.1021/cm402180t.
- Fındık, Volkan; Degirmenci, Isa; Çatak, Şaron; Aviyente, Viktorya (January 2019). "Theoretical investigation of thiol-ene click reactions: A DFT perspective". European Polymer Journal. 110: 211–220. doi:10.1016/j.eurpolymj.2018.11.030.
- Sy Piecco, Kurt W. E.; Aboelenen, Ahmed M.; Pyle, Joseph R.; Vicente, Juvinch R.; Gautam, Dinesh; Chen, Jixin (2018). "Kinetic Model under Light-Limited Condition for Photoinitiated Thiol–Ene Coupling Reactions". ACS Omega. 3 (10): 14327–14332. doi:10.1021/acsomega.8b01725. PMC 6210074. PMID 30411064.
- Lynch, Dylan M.; Scanlan, Eoin M. (2020-07-07). "Thiyl Radicals: Versatile Reactive Intermediates for Cyclization of Unsaturated Substrates". Molecules. 25 (13): 3094. doi:10.3390/molecules25133094. ISSN 1420-3049.
- Harrowven, David C; Lucas, Matthew C; Howes, Peter D (June 1999). "Total syntheses of aplysin and debromoaplysin using a diastereoselective, sulfur mediated radical cyclisation strategy". Tetrahedron Letters. 40 (23): 4443–4444. doi:10.1016/S0040-4039(99)00768-6.
- Miyata, Okiko; Ozawa, Yoshiki; Ninomiya, Ichiya; Naito, Takeaki (August 2000). "Radical Cyclization in Heterocycle Synthesis. Part 10: A Concise Synthesis of (−)-Kainic Acid via Sulfanyl Radical Addition–Cyclization–Elimination Reaction". Tetrahedron. 56 (34): 6199–6207. doi:10.1016/S0040-4020(00)00579-2.
- Crick, Peter J.; Simpkins, Nigel S.; Highton, Adrian (2011-12-16). "Synthesis of the Asperparaline Core by a Radical Cascade". Organic Letters. 13 (24): 6472–6475. doi:10.1021/ol202769f. ISSN 1523-7060.
- Keck, Gary E.; Wager, Travis T.; Rodriquez, J. Felix Duarte (June 1999). "Total Syntheses of (−)-Lycoricidine, (+)-Lycoricidine, and (+)-Narciclasine via 6- exo Cyclizations of Substituted Vinyl Radicals with Oxime Ethers †". Journal of the American Chemical Society. 121 (22): 5176–5190. doi:10.1021/ja9826688. ISSN 0002-7863.
- Scanlan, Eoin; Corcé, Vincent; Malone, Aoife (19 November 2014). "Synthetic Applications of Intramolecular Thiol-Ene "Click" Reactions". Molecules. 19 (11): 19137–19151. doi:10.3390/molecules191119137. PMC 6271571. PMID 25415476.
- Walling, Cheves; Helmreich, Wolf (March 1959). "Reactivity and Reversibility in the Reaction of Thiyl Radicals with Olefins". Journal of the American Chemical Society. 81 (5): 1144–1148. doi:10.1021/ja01514a032.
- Killops, Kato L.; Campos, Luis M.; Hawker, Craig J. (April 2008). "Robust, Efficient, and Orthogonal Synthesis of Dendrimers via Thiol-ene "Click" Chemistry". Journal of the American Chemical Society. 130 (15): 5062–5064. CiteSeerX 10.1.1.658.8715. doi:10.1021/ja8006325. PMID 18355008.
- Montañez, Maria I.; Campos, Luis M.; Antoni, Per; Hed, Yvonne; Walter, Marie V.; Krull, Brandon T.; Khan, Anzar; Hult, Anders; Hawker, Craig J.; Malkoch, Michael (27 July 2010). "Accelerated Growth of Dendrimers via Thiol−Ene and Esterification Reactions". Macromolecules. 43 (14): 6004–6013. Bibcode:2010MaMol..43.6004M. CiteSeerX 10.1.1.661.7797. doi:10.1021/ma1009935.
- Rissing, Christiana; Son, David Y. (8 June 2009). "Application of Thiol−Ene Chemistry to the Preparation of Carbosilane−Thioether Dendrimers". Organometallics. 28 (11): 3167–3172. doi:10.1021/om9001395.
- Son, D. Y.; Rissing, C.; Chen, L.; Andersson, T. E. (2010). "Thiol-ene Chemistry for the Synthesis and Modification of Branched Organosilicon Polymers". Polymer Preprints. 51 (2): 730–731.
- Jonkheijm, Pascal; Weinrich, Dirk; Köhn, Maja; Engelkamp, Hans; Christianen, Peter C. M.; Kuhlmann, Jürgen; Maan, Jan C.; Nüsse, Dirk; Schroeder, Hendrik; Wacker, Ron; Breinbauer, Rolf; Niemeyer, Christof M.; Waldmann, Herbert (26 May 2008). "Photochemical Surface Patterning by the Thiol-Ene Reaction". Angewandte Chemie International Edition. 47 (23): 4421–4424. doi:10.1002/anie.200800101. PMID 18428169.
- Kurt Waldo E. Sy Piecco, Juvinch R. Vicente, Joseph R. Pyle, David C. Ingram, Martin E. Kordesch, Jixin Chen (2019). "Reusable Chemically-Micropatterned Substrates via Sequential Photoinitiated Thiol-Ene Reactions as Template for Perovskite Thin-Film Microarrays". ChemRxiv.CS1 maint: uses authors parameter (link)
- Shafagh, Reza; Vastesson, Alexander; Guo, Weijin; van der Wijngaart, Wouter; Haraldsson, Tommy (2018). "E-Beam Nanostructuring and Direct Click Biofunctionalization of Thiol–Ene Resist". ACS Nano. 12 (10): 9940–9946. doi:10.1021/acsnano.8b03709. PMID 30212184.