Trimethylenemethane cycloaddition
Trimethylenemethane cycloaddition is the formal [3+2] annulation of trimethylenemethane (TMM) derivatives to two-atom pi systems. Although TMM itself is too reactive and unstable to be stored, reagents which can generate TMM or TMM synthons in situ can be used to effect cycloaddition reactions with appropriate electron acceptors. Generally, electron-deficient pi bonds undergo cyclization with TMMs more easily than electron-rich pi bonds.[1]
Introduction
Trimethylenemethane is a neutral, four-carbon molecule composed of four pi bonds; thus, it must be expressed either as a non-Kekulé molecule or a zwitterion. The orbital energy levels of TMM reveal that it possesses singlet and triplet states; generally, these states exhibit different reactivity and selectivity profiles.[2] A singlet [3+2] cycloaddition, when it is concerted, is believed to proceed under frontier orbital control. When electron-rich TMMs are involved, the A orbital serves as the HOMO (leading to fused products if the TMM is cyclic). When electron-poor (or unsubstituted) TMMs are involved, the S orbital serves as the HOMO (leading to bridged products if the TMM is cyclic). Cycloadditions involving the triplet state are stepwise, and usually result in configurational scrambling in the two-atom component.[3]

The rapid closure of TMMs to methylenecyclopropenes (MCPs) is a general problem that affects the rate and yield of [3+2] cycloaddition reactions involving this class of reaction intermediates.[4] The problem is generally less severe for five-membered, cyclic TMMs due to ring strain in the corresponding MCPs. When ring closure and TMM dimerization can be controlled, [3+2] cycloaddition affords isomeric mixtures of methylenecyclopentanes. Three classes of compounds have been used to generate synthetically useful TMM intermediates: diazenes, silyl-substituted allylic acetates and methylenecyclopropenes. Transition metal catalysis can be used with the latter two classes, although polar MCPs may open under light or heat (see below).
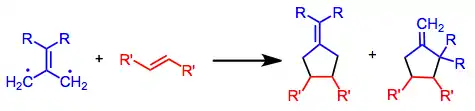
Mechanism and stereochemistry
Prevailing mechanisms
Diazenes may extrude nitrogen to provide discrete TMM intermediates. Generally, bridged diazenes are used to avoid competitive closure to MCPs and dimerization reactions.[5] In combination with an alkenic acceptor, cyclization to either fused or bridged products takes place. Fused products are generally favored, unless the diazene precursor is substituted with electron-donating groups at the methylene carbon atom. The configuration of the alkene is maintained as long as the reaction is proceeding through a singlet TMM.[6]

When stabilizing groups are present, MCPs may open to the corresponding zwitterionic TMMs.[7] Acetal 1 has been used in this context, and provides cyclopentanes with the acetal functionality exo to the newly formed ring with high selectivity. This reaction is also stereospecific with respect to alkene geometry, and exhibits high selectivity for endo products in most cases.

MCPs lacking stabilizing groups may generate TMM synthons in the presence of palladium(0) or nickel(0) catalysts.[8] Formal insertion of the catalyst into either of the two chemically distinct cyclopropane bonds (called "distal" and "proximal" to reflect their distance from the double bond) has the potential to generate isomeric products. Generally, palladium catalysts cause formal distal bond cleavage. This process is believed to occur through direct attack of the distal bond on the coordinated alkene. The reaction is stepwise and lacks stereospecificity under both palladium and nickel catalysis.

Silylated allylic acetates, carbonates and other substituted allyl compounds may form TMM synthons under palladium catalysis.[9] The reaction is highly regioselective, providing only the substitution pattern shown below regardless of the position of the R' group on the starting allylic acetate. However, cyclization takes place in a stepwise fashion and does not exhibit stereospecificity. Rapid racemization of chiral pi-allyl palladium complexes occurs, and only moderate diastereoselectivity is observed in reactions of chiral allylic acetates. Chiral non-racemic alkenes, however, may exhibit moderate to high diastereoselectivity.

Stereoselective variants
Chiral auxiliaries on the alkene partner have been used for stereoselective transformations. In the reaction of camphorsultam-derived unsaturated amides, lower temperatures were needed to achieve high selectivities.[10]
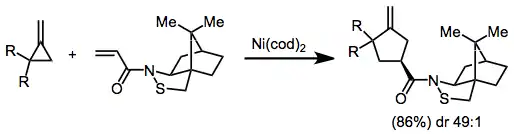
In reactions of silyl-substituted allylic acetates, chiral sulfoxides can be used to enforce high diastereofacial selectivity.[11]

Scope and limitations
The primary limitations of TMM cycloadditions employing diazenes are competitive MCP and dimer formation. To circumvent these problems, either very high concentrations of alkene must be used or the cycloaddition must be intramolecular. Stereoselectivity and site selectivity may also be higher in intramolecular variants of cycloadditions starting from diazenes.[12]

Usually, unless a cyclic pi system is involved TMM cycloadditions exhibit 2π periselectivity and do not react with larger pi systems. Polar MCPs, for example, react only with the 2,3 double bond of polyunsaturated esters.[13]

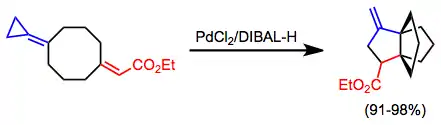
Transition-metal catalyzed reactions have the potential to quickly generate an interesting functionality. Propellanes have been generated from intramolecular cyclization under palladium catalysis.[14]
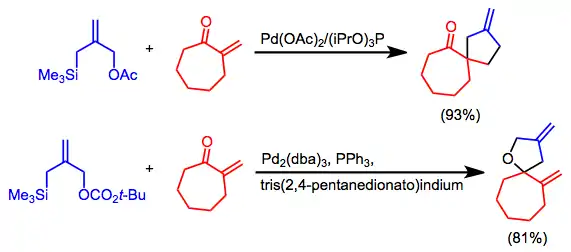
Silylated allylic acetates may be employed for intra- or intermolecular applications. Carbonyl compounds may be used as the 2π component under the appropriate conditions. For instance, in the presence of an indium co-catalyst, the reactive 2π component of the cycloaddition below switches from the C-C to the C-O double bond.[15]
Polarized trimethylenemethanes generated from polar MCPs are also useful substrates for [3+2] reactions with polar double bonds as the 2π component. Orthoester products are generally favored over ketene acetals.[16]

Synthetic applications
The high stereospecificity and stereoselectivity inherent in many TMM cycloaddition reactions is a significant advantage; for instance, the trans ring junction in TMM cycloaddition adduct 2 was carried through in a synthesis of (+)-brefeldin A.[17]

Comparison with other methods
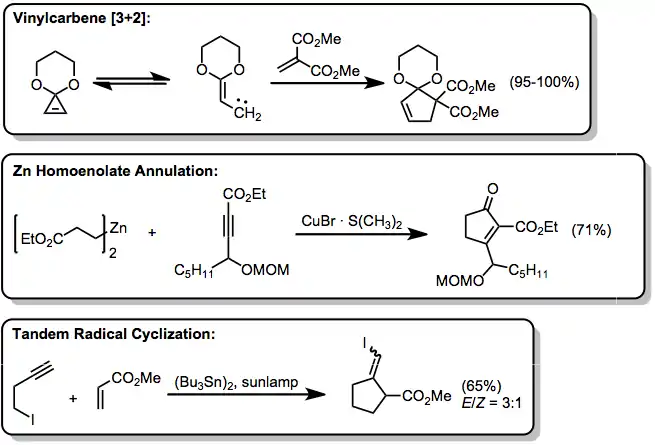
Although 1,3-dipolar cycloaddition is a useful method for the generation of five-membered heterocyclic compounds, few methods exist to synthesize five-membered carbocyclic rings in a single step via annulation. Most of these, like TMM cycloaddition, rely on the generation of a suitable three-atom component for combination with a stable two-atom partner such as an alkene or alkyne. When heated, cyclopropene acetals rearrange to vinylcarbenes, which can serve as the three-atom component in cycloadditions with highly electron-deficient alkenes.[18] Zinc homoenolates can also serve as indirect three-atom components, and undergo cyclization to cyclopentenones in the presence of an unsaturated ester.[19] Tandem intermolecular-intramolecular cyclization of homopropargylic radicals leads to methylenecyclopropanes.[20]
Experimental conditions and procedure
Typical conditions
The optimal conditions for TMM cycloadditions depend on both the TMM source and two-atom component. However, a few general principles for each of the TMM sources have emerged.
Reactions of diazenes should employ degassed solvents to avoid radical reactions with oxygen. Tetrahydrofuran (THF) at reflux is the most commonly employed solvent system, but photodissociation conditions at low temperature may also be used.
Reactions employing polar MCPs are usually carried out in a polar solvent to facilitate formation of the TMM intermediate. Although rigorous exclusion of air and water is not required, it is generally preferred.
For transition-metal catalyzed MCP reactions, the choice of catalyst and ligand system is key. Generally, phosphine or phosphite ligands are required in conjunction with a palladium(0) or nickel(0) source; the most common are Pd2(dba)3 and Ni(cod)2. Tri(isopropyl)phosphine is the most common ligand used with palladium, and triarylphosphites are usually added in nickel-catalyzed reactions.
For transition-metal catalyzed reactions of silylated allylic acetates, the most commonly used catalyst system is palladium(II) acetate and tri(isopropyl)phosphite. Reactions are usually carried out in THF at temperatures ranging from 60 to 110 °C. The choice of solvent or leaving group may affect the course of the reaction.
References
- Yamago, S.; Nakamura, E. Org. React. 2003, 61, 1. doi:10.1002/0471264180.or061.01
- Baseman, R. J.; Pratt, D. W.; Chow, M.; Dowd, P. J. Am. Chem. Soc. 1976, 98, 5726.
- Platz, M. S.; Berson, J. A. J. Am. Chem. Soc. 1980, 102, 2358.
- Yamago, S.; Nakamura, E. J. Am. Chem. Soc. 1989, 111, 7285.
- Berson, J. A. Acc. Chem. Res. 1978, 11, 446.
- Berson, J. A.; Duncan, C. D.; Corwin, L. R. J. Am. Chem. Soc. 1974, 96, 6175.
- Nakamura, E.; Yamago, S.; Ejiri, S.; Dorigo, A. E.; Morokuma, K. J. Am. Chem. Soc. 1991, 113, 3183.
- Binger, P.; Büch, H. M. Top. Curr. Chem. 1987, 135, 77.
- Trost, B. M. Angew. Chem. Int. Ed. Engl. 1986, 25, 1.
- Binger, P.; Schäfer, B. Tetrahedron Lett. 1988, 29, 529.
- Chaigne, F.; Gotteland, J.-P.; Malacria, M. Tetrahedron Lett. 1989, 30, 1803.
- Little, R. D.; Muller, G. W. J. Am. Chem. Soc. 1981, 103, 2744.
- Nakamura, E.; Yamago, S. Acc. Chem. Res. 2002, 35, 867.
- Yamago, S.; Nakamura, E. Tetrahedron 1989, 45, 3081.
- Trost, B. M.; Sharma, S.; Schmidt, T. J. Am. Chem. Soc. 1992, 114, 7903.
- Yamago, S.; Nakamura, E. J. Org. Chem. 1990, 55, 5553.
- Trost, B. M.; Lynch, J.; Renaut, P.; Steinman, D. H. J. Am. Chem. Soc. 1986, 108, 284.
- Boger, D. L.; Brotherton, C. E. J. Am. Chem. Soc. 1986, 108, 6695.
- Crimmins, M. T.; Nantermet, P. G. J. Org. Chem. 1990, 55, 4235.
- Curran, D. P.; Chen, M.-H. J. Am. Chem. Soc. 1987, 109, 6558.