Tumor metabolome
The study of the tumor metabolism, also known as tumor metabolome describes the different characteristic metabolic changes in tumor cells. The characteristic attributes[2] of the tumor metabolome are high glycolytic enzyme activities, the expression of the pyruvate kinase isoenzyme type M2, increased channeling of glucose carbons into synthetic processes, such as nucleic acid, amino acid and phospholipid synthesis, a high rate of pyrimidine and purine de novo synthesis, a low ratio of Adenosine triphosphate and Guanosine triphosphate to Cytidine triphosphate and Uridine triphosphate, low Adenosine monophosphate levels, high glutaminolytic capacities, release of immunosuppressive substances and dependency on methionine.
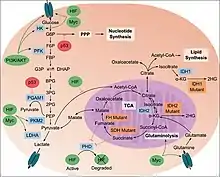
Although the link between the cancer and metabolism was observed in the early days of cancer research by Otto Heinrich Warburg,[3] which is also known as Warburg hypothesis, not much substantial research was carried out until the late 1990s because of the lack of in vitro tumor models and the difficulty in creating environments that lack oxygen. Recent research has revealed that metabolic reprogramming occurs as a consequence of mutations in cancer genes and alterations in cellular signaling. Therefore, the alteration of cellular and energy metabolism has been suggested as one of The Hallmarks of Cancer.[4][5]
Warburg effect and glycolysis
High amount of aerobic glycolysis (also known as the Warburg effect) distinguishes cancer cells from normal cells. The conversion of glucose to lactate rather than metabolizing it in the mitochondria through oxidative phosphorylation, (which can also occur in hypoxic normal cells) persists in malignant tumor despite the presence of oxygen. This process normally inhibits glycolysis which is also known as Pasteur effect. One of the reasons it is observed is because of the malfunction of mitochondria. Although ATP production by glycolysis can be more rapid than by oxidative phosphorylation, it is far less efficient in terms of ATP generated per unit of glucose consumed. Rather than oxidizing glucose for ATP production, glucose in cancer cells tends to be used for anabolic processes, such as ribose production, protein glycosylation and serine synthesis. This shift therefore demands that tumor cells implement an abnormally high rate of glucose uptake to meet their increased needs.[5]
As neoplastic cells accumulate in three-dimensional multicellular masses, local low nutrient and oxygen levels trigger the growth of new blood vessels into the neoplasm. The imperfect neovasculature in the tumor bed is poorly formed and is inefficient. It therefore, causes nutrient and hypoxic stress (or a state of hypoxia).[6][7] In this regard, cancer cells and stromal cells can symbiotically recycle and maximize the use of nutrients. Hypoxic adaptation by cancer cells is essential for survival and progression of a tumor.[8][9] In addition to cell-autonomous changes that drive a cancer cell to proliferate and contribute to tumorigenesis, it has also been observed that alterations in whole-organism metabolism such as obesity are associated with heightened risks for a variety of cancers.[10]
Role of signalling pathway in cancer metabolism
In the PI3K/AKT/mTOR pathway, AKT1 (also known as Protein Kinase B or PKB) is an important driver of the tumor glycolytic phenotype and stimulates ATP generation. AKT1 stimulates glycolysis by increasing the expression and membrane translocation of glucose transporters and by phosphorylating key glycolytic enzymes, such as hexokinase and phosphofructokinase 2. This leads to inhibition of forkhead box subfamily O transcription factors, leading to the increase of glycolytic capacity. Activated mTOR stimulates protein and lipid biosynthesis and cell growth in response to sufficient nutrient and energy conditions and is often constitutively activated during tumorigenesis.[5] mTOR directly stimulates mRNA translation and ribosome biogenesis, and indirectly causes other metabolic changes by activating transcription factors such as hypoxia-inducible factor 1 (HIF1A). The subsequent HIF1-dependent metabolic changes are a major determinant of the glycolytic phenotype downstream of PI3K, AKT1 and mTOR.[11]
Role of tumor suppressor and oncogenes
Apart from being as a general tumor suppressor gene, p53 also plays an important part in regulating of metabolism. p53 activates hexokinase 2 (HK2) that converts glucose to glucose-6-phosphate (G6P) which enters glycolysis to produce ATP, or enters the pentose phosphate pathway (PPP). It therefore, supports macromolecular biosynthesis by producing reducing potential in the form of reduced Nicotinamide adenine dinucleotide phosphate (NADPH) and/or ribose that are used for nucleotide synthesis.[12] p53 inhibits the glycolytic pathway by upregulating the expression of TP53-induced glycolysis and apoptosis regulator. Wild-type p53 supports the expression of PTEN (gene), which inhibits the PI3K pathway, thereby suppressing glycolysis. POU2F1 also cooperate with p53 in regulating the balance between oxidative and glycolytic metabolism. It provides resistance to oxidative stress that would regulates a set of genes that increase glucose metabolism and reduce mitochondrial respiration. This will provide additive force when the p53 is lost.[5] Mutated Ras also enhances glycolysis, partly through increasing the activity of Myc and hypoxia-inducible factors. Although HIF-1 inhibits Myc, HIF-2 activates Myc causing the multiplicity of the tumor cells.[9]
TCA cycle in cancer metabolism
Mutations in fumarate hydratase are found among patients suffering from kidney cancers, and mutations in succinate dehydrogenase were found in patients with pheochromocytoma and paragangliomas. These mutations cause a disruption of the TCA cycle with the accumulation of fumarate or succinate, both of which can inhibit dioxygenases or prolyl hydrolases that mediate the degradation of HIF proteins. HIF-1 could be elevated under aerobic conditions downstream from activated PI3K, which stimulates the synthesis of HIF-1. Loss of the tumor suppressor VHL in kidney cancer also stabilizes HIF-1, permitting it to activate glycolytic genes, which are normally activated by HIF-1 under hypoxic conditions.[9] HIF1 then would activate the pyruvate dehydrogenase kinase (PDKs), which inactivate the mitochondrial pyruvate dehydrogenase complex. It reduces the flow of glucose-derived pyruvate into the tricarboxylic acid (citric acid cycle or TCA cycle). This reduction in pyruvate flux into the TCA cycle decreases the rate of oxidative phosphorylation and oxygen consumption, reinforcing the glycolytic phenotype and sparing oxygen under hypoxic conditions.[13][14]
M2 isoform of pyruvate kinase
Pyruvate kinase type M2 or PKM2 is present in embryonic, adult stem cells. It is also expressed by many tumor cells. The alterations to metabolism by PKM2 increases ATP resources, stimulates macromolecular biosynthesis and redox control. Pyruvate kinase catalyses the ATP-generating step of glycolysis in which phosphoenolpyruvate (PEP) is converted to pyruvate. This is a rate-limiting step.[15] It decreases the glycolysis activity and allows carbohydrate metabolites to enter other pathways, like hexosamine pathway, Uridine diphosphate glucose–glucose synthesis, glycerol synthesis and Pentose phosphate pathway or PPP. It helps in generating macromolecule precursors, that are necessary to support cell proliferation, and reducing equivalents such as NADPH.[16][17] It has been observed in some studies that MYC promotes expression of PKM2 over PKM1 by modulating exon splicing.[5]
A key molecule produced as a result of the oxidative PPP by PKM2 is NADPH. NADPH functions as a cofactor and provides reducing power in many enzymatic reactions that are crucial for macromolecular biosynthesis. Another mechanism by which NADPH is produced in mammalian cells is the reaction converting isocitrate to α-ketoglutarate (αKG), which is catalysed by NADP-dependent isocitrate dehydrogenase 1 (IDH1) and IDH2 and have been found linked to tumorigenesis in glioblastoma and acute myeloid leukemia.[18][19] They are also found to interact with arginine residues required for isocitrate binding in the active site of the proteins of IDH1 and IDH2.[5]
Fatty acid synthesis
Fatty acid synthesis is an anabolic process that starts from the conversion of acetyl-CoA to malonyl-CoA by acetyl-CoA carboxylase. Malonyl CoA leads to fatty acid synthesis (FAS) and is involved in the elongation of fatty acids through Fatty acid synthase (FASN). Although aerobic glycolysis is the best documented metabolic phenotype of tumor cells, it is not a universal feature of all human cancers. Amino acids and fatty acids have been shown to function as fuels for tumor cells to proliferate. The carnitine palmitoyltransferase enzymes that regulate the β-oxidation of fatty acids may have a key role in determining some of these phenotypes.[5] Enhanced fatty acid synthesis provides lipids for membrane biogenesis to tumor cells and hence, it gives advantage in both growth and survival of the cell.
Adaptation and resistance to drugs
It has also been seen that metabolic phenotype of tumor cells changes to adapt to the prevailing local conditions. Some of the fatty acids have been linked to acquire resistance against some of the cancer drugs. Fatty acid synthase (FASN), a key complex catalyzing fatty acid synthesis has been found to be linked to acquired docetaxel, trastuzumab and adriamycin resistance in breast cancer. Similar resistance have been found with intrinsic gemcitabine and radiation resistance in pancreatic cancer. Glutaminolysis is linked to cisplatin resistance via the activation of mTORC1 signaling in gastric cancer.[20]
Metabolic biomarkers of tumors
NADPH plays an important role as an antioxidant by decreasing the reactive oxygen produced during rapid cell proliferation. It has been shown that attenuation of the PPP would dampen NADPH production in cancer cells, leading to the decrease in macromolecular biosynthesis and rendering the transformed cells that are vulnerable free radical-mediated damage. In this way, the advantage conferred by PKM2 expression would be eliminated. In preclinical studies, drugs such as 6-amino-nicotinamide (6-AN), which inhibits G6P dehydrogenase, the enzyme that initiates the PPP have shown anti-tumorigenic effects in leukemia, glioblastoma and lung cancer cell lines.[21]
Cyclosporine inhibits TOR and is used as an effective immunosuppressant. Mycophenolic acid inhibits of IMPDH and pyrimidine biosynthesis and is clinically used as immunosuppressant. Both agents also display anti-tumor effects in animal studies.[9] Metabolites such as Alanine, Saturated lipids, Glycine, Lactate, Myo-Inositol, Nucleotides, Polyunsaturated fatty acids and Taurine are considered as the potential biomarkers in various studies.[22]
Glutaminolysis
The use of the amino acid glutamine as an energy source is facilitated by the multistep catabolism of glutamine called glutaminolysis. This energy pathway is upregulated in cancer, which may represent a therapeutic target as cancer cells are thought to be more dependent on glutamine than healthy cells.[23] This especially holds true for specific tumor types that are metabolically dysregulated, such as malignant brain tumors (i.e. glioblastoma) that carry mutations in the IDH1 gene. These tumors use glutamine or the structurally related amino acid glutamate as an energy source and a chemotactic sensor in the brain, which increases their malignancy and may explain why these tumors grow so invasive.[9][10]
References
- Vermeersch, Kathleen A.; Styczynski, Mark P. (2013). "Applications of metabolomics in cancer research". J. Carcinog. 12 (9): 9. doi:10.4103/1477-3163.113622. PMC 3709411. PMID 23858297.
- Mazurek, S.; Eigenbrodt E. (March–April 2003). "The tumor metabolome". Anticancer Research. 23 (2A): 1149–1154. PMID 12820363.
- Warburg O (1956). "On the origin of cancer cells". Science. 123 (3191): 309–314. doi:10.1126/science.123.3191.309. PMID 13298683.
- Hanahan, Douglas; Weinberg, Robert A. (March 2011). "Hallmarks of Cancer: The Next Generation". Cell. 144 (5): 646–674. doi:10.1016/j.cell.2011.02.013. PMID 21376230.
- Cairns, Rob A.; Harris, Isaac S.; Mak, Tak W. (2011). "Regulation of cancer cell metabolism". Nat Rev Cancer. 11 (2): 85–95. doi:10.1038/nrc2981. PMID 21258394. S2CID 8891526.
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E, et al. (1998). "Role of HIF-1 alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis". Nature. 394 (6692): 485–490. doi:10.1038/28867. PMID 9697772. S2CID 4419118.
- Semenza, G.L. (2012). "Hypoxia-inducible factors in physiology and medicine". Cell. 148 (3): 399–408. doi:10.1016/j.cell.2012.01.021. PMC 3437543. PMID 22304911.
- Semenza, G.L. (2010). "HIF-1: Upstream and downstream of cancer metabolism". Curr Opin Genet Dev. 20 (1): 51–56. doi:10.1016/j.gde.2009.10.009. PMC 2822127. PMID 19942427.
- Dang, Chi V. (2012). "Links between metabolism and cancer". Genes & Development. 26 (9): 877–890. doi:10.1101/gad.189365.112. PMC 3347786. PMID 22549953.
- Khandekar, MJ; Cohen P.; Spiegelman BM. (2011). "Molecular mechanisms of cancer development in obesity". Nat Rev Cancer. 11 (12): 886–895. doi:10.1038/nrc3174. PMID 22113164. S2CID 1978204.
- Guertin, D. A; Sabatini, D. M. (January 2007). "Defining the role of mTOR in cancer". Cancer Cell. 12 (1): 9–22. doi:10.1016/j.ccr.2007.05.008. PMID 17613433.
- Mathupala, S. P.; Heese, C.; Pedersen, P. L. (1997). "Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53". J. Biol. Chem. 272 (36): 22776–22780. doi:10.1074/jbc.272.36.22776. PMID 9278438.
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006). "HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption". Cell Metabolism. 3 (3): 187–197. doi:10.1016/j.cmet.2006.01.012. PMID 16517406.
- Kim, J. W.; Tchernyshyov, I.; Semenza, G. L.; Dang, C. V. (2006). "HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia". Cell Metabolism. 3 (3): 177–185. doi:10.1016/j.cmet.2006.02.002. PMID 16517405.
- Mazurek, S.; Boschek, C. B.; Hugo, F.; Eigenbrodt, E. (2005). "Pyruvate kinase type M2 and its role in tumor growth and spreading". Semin. Cancer Biol. 15 (4): 300–308. doi:10.1016/j.semcancer.2005.04.009. PMID 15908230.
- Vander Heiden, M. G.; Cantley, L. C.; Thompson, C. B. (2009). "Understanding the Warburg effect: the metabolic requirements of cell proliferation". Science. 324 (5930): 1029–1033. doi:10.1126/science.1160809. PMC 2849637. PMID 19460998.
- Fang, M.; et al. (2010). "The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway". Cell. 143 (5): 711–724. doi:10.1016/j.cell.2010.10.010. PMID 21074248. S2CID 11891493.
- Parsons, D. W.; et al. (2008). "An integrated genomic analysis of human glioblastoma multiforme". Science. 321 (5897): 1807–1812. doi:10.1126/science.1164382. PMC 2820389. PMID 18772396.
- Mardis, E. R.; et al. (2009). "Recurring mutations found by sequencing an acute myeloid leukemia genome". N. Engl. J. Med. 361 (11): 1058–1066. doi:10.1056/NEJMoa0903840. PMC 3201812. PMID 19657110.
- Zhao, Y; et al. (2013). "Targeting cellular metabolism to improve cancer therapeutics". Cell Metabolism. 4 (3): e532. doi:10.1038/cddis.2013.60. PMC 3613838. PMID 23470539.
- Budihardjo, I. I; et al. (1998). "6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin". Clin. Cancer Res. 4 (1): 117–130. PMID 9516960.
- Griffin, Julian L.; Shockcor, John P. (2004). "Metabolic profiles of cancer cells". Nature Reviews Cancer. 4 (7): 551–561. doi:10.1038/nrc1390. PMID 15229480. S2CID 527894.
- Chen, JQ; Russo, J (December 2012). "Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1826 (2): 370–84. doi:10.1016/j.bbcan.2012.06.004. PMID 22750268.