Wolff–Kishner reduction
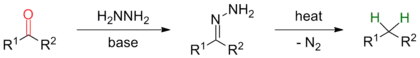
The Wolff–Kishner reduction is a reaction used in organic chemistry to convert carbonyl functionalities into methylene groups. In the context of complex molecule synthesis, it is most frequently employed to remove a carbonyl group after it has served its synthetic purpose of activating an intermediate in a preceding step. As such, there is no obvious retron for this reaction. Originally reported by Nikolai Kischner in 1911[1] and Ludwig Wolff in 1912,[2] it has been applied to the total synthesis of scopadulcic acid B,[3] aspidospermidine[4][5] and dysidiolide.[6]
| Wolff-Kishner reduction | |
|---|---|
| Named after | Ludwig Wolff Nikolai Kischner |
| Reaction type | Organic redox reaction |
| Identifiers | |
| Organic Chemistry Portal | wolff-kishner-reduction |
| RSC ontology ID | RXNO:0000226 |
In general, the reaction mechanism first involves the in situ generation of a hydrazone by condensation of hydrazine with the ketone or aldehyde substrate. Sometimes it is however advantageous to use a pre-formed hydrazone as substrate (see modifications). The rate determining step of the reaction is de-protonation of the hydrazone by an alkoxide base to form a diimide anion by a concerted, solvent mediated protonation/de-protonation step. Collapse of this alkyldiimide with loss of N2 leads to formation of an alkylanion which can be protonated by solvent to give the desired product.
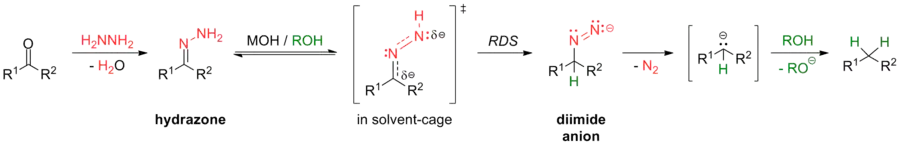
Because the Wolff–Kishner reduction requires highly basic conditions, it is unsuitable for base-sensitive substrates. In some cases, formation of the required hydrazone will not occur at sterically hindered carbonyl groups, preventing the reaction. However, this method can be superior to the related Clemmensen reduction for compounds containing acid-sensitive functional groups such as pyrroles and for high-molecular weight compounds.
History
The Wolff–Kishner reduction was discovered independently by N. Kishner[1] in 1911 and Ludwig Wolff in 1912.[2] Kishner found that addition of pre-formed hydrazone to hot potassium hydroxide containing crushed platinized porous plate led to formation of the corresponding hydrocarbon. A review titled “Disability, Despotism, Deoxygenation—From Exile to Academy Member: Nikolai Matveevich Kizhner” describing the life and work of Kishner was published in 2013.[7]

Wolff later accomplished the same result by heating an ethanol solution of semicarbazones or hydrazones in a sealed tube to 180 °C in the presence of sodium ethoxide.

The method developed by Kishner has the advantage of avoiding the requirement of a sealed tube, but both methodologies suffered from unreliability when applied to many hindered substrates. These disadvantages promoted the development of Wolff’s procedure, wherein the use of high-boiling solvents such as ethylene glycol and triethylene glycol were implemented to allow for the high temperatures required for the reaction while avoiding the need of a sealed tube.[8][9] These initial modifications were followed by many other improvements as described below.
Mechanism
The mechanism of the Wolff–Kishner reduction has been studied by Szmant and coworkers.[10][11][12][13] According to Szmant's research, the first step in this reaction is the formation of a hydrazone anion 1 by deprotonation of the terminal nitrogen by MOH. If semicarbazones are used as substrates, initial conversion into the corresponding hydrazone is followed by deprotonation.[2] A range of mechanistic data suggests that the rate-determining step involves formation of a new carbon–hydrogen bond at the carbon terminal in the delocalized hydrazone anion. This proton capture takes place in a concerted fashion with a solvent-induced abstraction of the second proton at the nitrogen terminal. Szmant’s finding that this reaction is first order in both hydroxide ion and ketone hydrazone supports this mechanistic proposal.[14] Several molecules of solvent have to be involved in this process in order to allow for a concerted process. A detailed Hammett analysis[10] of aryl aldehydes, methyl aryl ketones and diaryl ketones showed a non-linear relationship which the authors attribute to the complexity of the rate-determining step. Mildly electron-withdrawing substituents favor carbon-hydrogen bond formation, but highly electron-withdrawing substituents will decrease the negative charge at the terminal nitrogen and in turn favor a bigger and harder solvation shell that will render breaking of the N-H bond more difficult. The exceptionally high negative entropy of activation values observed can be explained by the high degree of organization in the proposed transition state.
It was furthermore found that the rate of the reaction depends on the concentration of the hydroxylic solvent and on the cation in the alkoxide catalyst. The presence of crown ether in the reaction medium can increase the reactivity of the hydrazone anion 1 by dissociating the ion pair and therefore enhance the reaction rate.[13] The final step of the Wolff–Kishner reduction is the collapse of the diimide anion 2 in the presence of a proton source to give the hydrocarbon via loss of dinitrogen to afford an alkyl anion 3, which undergoes rapid and irreversible acid-base reaction with solvent to give the alkane. Evidence for this high-energy intermediate was obtained by Taber via intramolecular trapping. The stereochemical outcome of this experiment was more consistent with an alkyl anion intermediate than the alternative possibility of an alkyl radical.[15] The overall driving force of the reaction is the evolution of nitrogen gas from the reaction mixture.

Modifications
Many of the efforts devoted to improve the Wolff–Kishner reduction have focused on more efficient formation of the hydrazone intermediate by removal of water and a faster rate of hydrazone decomposition by increasing the reaction temperature.[8][9] Some of the newer modifications provide more significant advances and allow for reactions under considerably milder conditions. The table shows a summary of some of the modifications that have been developed since the initial discovery.
| Original procedure[1][2] | Huang Minlon[16] | Barton[17] | Cram[18] | Henbest[19] | Caglioti[20] | Myers[21] | |
|---|---|---|---|---|---|---|---|
| Reagents | carbonyl compound, 100% H2NNH2, Na or NaOEt | carbonyl compound, 85% H2NNH2, KOH | carbonyl compound, anhydrous H2NNH2, Na | preformed hydrazone, KOtBu | preformed hydrazone, KOtBu | tosylhydrazone, hydride donor | carbonyl compound, 1,2-bis(tert-butyldimethylsilyl)- hydrazine, Sc(OTf)3, KOtBu |
| Solvent | high-boiling solvent, e.g. ethylene glycol | high-boiling solvent, e.g. ethylene glycol | high-boiling solvent, e.g. diethylene glycol | anh. DMSO | toluene | THF | DMSO |
| Temperature | 200 °C | 180–200 °C (after removal of water and excess hydrazine) | 210 °C | 25 °C | 111 °C | 66 °C | 25 °C |
| Advantages | single step procedure | reduced reaction times, higher temperatures can be reached, no need to use anh. hydrazine | allows decarbonylation of sterically hindered substrates | proceeds at room temperature | no slow addition of hydrazone necessary | mild reaction conditions, possible with a variety of reducing agents | very mild reaction conditions |
| Disadvantages | long reaction times (50–100 h) | distillation necessary | harsh reaction conditions | isolation of hydrazone and slow addition necessary | isolation of hydrazone necessary | isolation of tosylhydrazone necessary. hydride donor may act as base | synthesis of 1,2-bis(tert-butyldimethylsilyl)- hydrazine necessary |
| Functional group tolerance | does not tolerate esters, amides, halogens, cyano-, and nitro-groups | similar to original procedure | similar to original procedure | tolerates amides | higher tolerance of α-substituents that would undergo elimination and α,β-unsaturated enones that would undergo migration under original conditions | tolerates esters, amides, cyano-, nitro- and chloro-substituents with NaBH3CN as hydride source, does not tolerate primary bromo- and iodo-substituents | not reported |
Huang Minlon modification
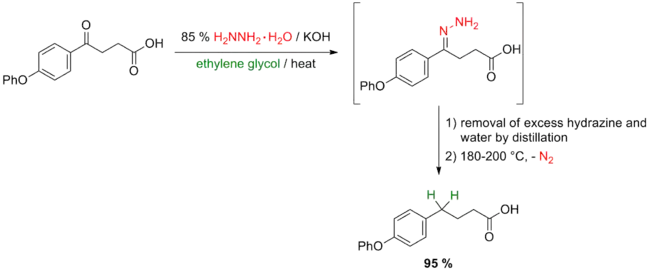
In 1946, Huang Minlon reported a modified procedure for the Wolff–Kishner reduction of ketones in which excess hydrazine and water were removed by distillation after hydrazone formation.[16][22] The temperature-lowering effect of water that was produced in hydrazone formation usually resulted in long reaction times and harsh reaction conditions even if anhydrous hydrazine was used in the formation of the hydrazone. The modified procedure consists of refluxing the carbonyl compound in 85% hydrazine hydrate with three equivalents of sodium hydroxide followed by distillation of water and excess hydrazine and elevation of the temperature to 200 °C. Significantly reduced reaction times and improved yields can be obtained using this modification. Huang Minlon's original report described the reduction of β-(p-phenoxybenzoyl)propionic acid to γ-(p-phenoxyphenyl)butyric acid in 95% yield compared to 48% yield obtained by the traditional procedure.
Barton modification
Nine years after Huang Minlon’s first modification, Barton developed a method for the reduction of sterically hindered carbonyl groups.[17] This method features vigorous exclusion of water, higher temperatures, and longer reaction times as well as sodium in diethylene glycol instead of alkoxide base. Under these conditions, some of the problems that normally arise with hindered ketones can be alleviated—for example, the C11-carbonyl group in the steroidal compound shown below was successfully reduced under Barton’s conditions while Huang–Minlon conditions failed to effect this transformation.
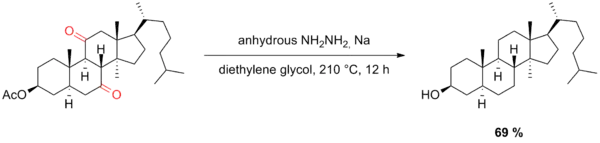
Cram modification
Slow addition of preformed hydrazones to potassium tert-butoxide in DMSO as reaction medium instead of glycols allows hydrocarbon formation to be conducted successfully at temperatures as low as 23 °C.[18] Cram attributed the higher reactivity in DMSO as solvent to higher base strength of potassium tert-butoxide in this medium.

This modification has not been exploited to great extent in organic synthesis due to the necessity to isolate preformed hydrazone substrates and to add the hydrazone over several hours to the reaction mixture.
Henbest modification
Henbest extended Cram’s procedure by refluxing carbonyl hydrazones and potassium tert-butoxide in dry toluene.[19] Slow addition of the hydrazone is not necessary and it was found that this procedure is better suited for carbonyl compounds prone to base-induced side reactions than Cram's modification. It has for example been found that double bond migration in α,β-unsaturated enones and functional group elimination of certain α-substituted ketones are less likely to occur under Henbest's conditions.[23]
Caglioti reaction
Treatment of tosylhydrazones with hydride-donor reagents to obtain the corresponding alkanes is known as the Caglioti reaction.[20][24] The initially reported reaction conditions have been modified and hydride donors such as sodium cyanoborohydride, sodium triacetoxyborohydride, or catecholborane can reduce tosylhydrazones to hydrocarbons.[25] The reaction proceeds under relatively mild conditions and can therefore tolerate a wider array of functional groups than the original procedure. Reductions with sodium cyanoborohydride as reducing agent can be conducted in the presence of esters, amides, cyano-, nitro- and chloro-substituents. Primary bromo- and iodo-substituents are displaced by nucleophilic hydride under these conditions.
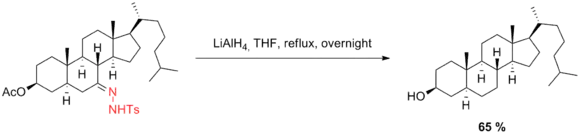
Several papers have studied the mechanism of this reduction and multiple reaction pathways are possible, depending on the pH of the reaction, the reducing agent used and the electronic properties of the substrate. [26][27] One possibility, occurring under acidic conditions, includes direct hydride attack of iminium ion 1 following prior protonation of the tosylhydrazone. The resulting tosylhydrazine derivative 2 subsequently undergoes elimination of p-toluenesulfinic acid and decomposes via a diimine intermediate 3 to the corresponding hydrocarbon.
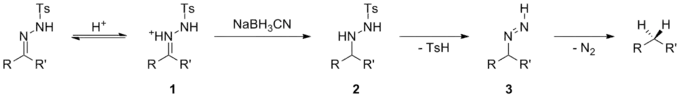
A slight variation of this mechanism occurs when tautomerization to the azohydrazone is facilitated by inductive effects. The transient azohydrazine 4 can then be reduced to the tosylhydrazine derivative 2 and furnish the decarbonylated product analogously to the first possibility. This mechanism operates when relatively weak hydride donors are used, such as sodium cyanoborohydride. It is known that these sodium cyanoborohydride is not strong enough to reduce imines, but can reduce iminium ions.

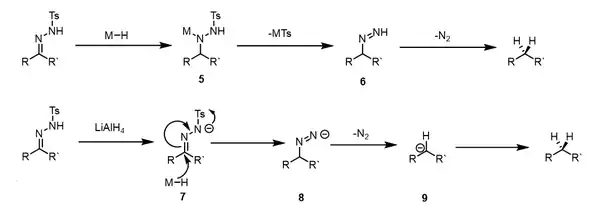
When stronger hydride donors are used, a different mechanism is operational, which avoids the use of acidic conditions. Hydride delivery occurs to give intermediate 5, followed by elimination of the metal sulfinate to give azo intermediate 6. This intermediate then decomposes, with loss of nitrogen gas, to give the reduced compound. When strongly basic hydride donors are used such as lithium aluminium hydride, then deprotonation of the tosyl hydrazone can occur before hydride delivery. Intermediate anion 7 can undergo hydride attack, eliminating a metal sulfinate to give azo anion 8. This readily decomposes to carbanion 9, which is protonated to give the reduced product.
As with the parent Wolff-Kishner reduction, the decarbonylation reaction can often fail due to unsuccessful formation of the corresponding tosylhydrazone. This is common for sterically hindered ketones, as was the case for the cyclic amino ketone shown below.[28]

Alternative methods of reduction can be employed when formation of the hydrazone fail, including thioketal reduction with Raney nickel or reaction with sodium triethylborohydride.
Deoxygenation of α,β-unsaturated carbonyl compounds
α,β-Unsaturated carbonyl tosylhydrazones can be converted into the corresponding alkenes with migration of the double bond. The reduction proceeds stereoselectively to furnish the E geometric isomer.[29]

A very mild method was developed by Kabalka et al. who used one equivalent of catecholborane to reduce α,β-unsaturated tosylhydrazones.[30]

Djerassi et al. studied the mechanism of NaBH3CN reduction of α,β-unsaturated tosylhydrazones. Based on deuterium-labeling experiments, they concluded that alkene formation is initiated by hydride reduction of the iminium ion followed by double bond migration and nitrogen extrusion which occur in a concerted manner.[31] Allylic diazene rearrangement as the final step in the reductive 1,3-transposition of α,β-unsaturated tosylhydrazones to the reduced alkenes can also be used to establish sp3-stereocenters from allylic diazenes containing prochiral stereocenters. The influence of the alkoxy stereocenter results in diastereoselective reduction of the α,β-unsaturated tosylhydrazone.[32] The authors predicted that diastereoselective transfer of the diazene hydrogen to one face of the prochiral alkene could be enforced during the suprafacial rearrangement.

Myers modification
In 2004, Myers and coworkers developed a method for the preparation of N-tert-butyldimethylsilylhydrazones from carbonyl-containing compounds.[21] These products can be used as a superior alternative to hydrazones in the transformation of ketones into alkanes. The advantages of this procedure are considerably milder reaction conditions and higher efficiency as well as operational convenience. The condensation of 1,2-bis(tert-butyldimethylsilyl)-hydrazine with aldehydes and ketones with Sc(OTf)3 as catalyst is rapid and efficient at ambient temperature. Formation and reduction of N-tert-butyldimethylsilylhydrazones can be conducted in a one pot procedure in high yield.

The newly developed method was compared directly to the standard Huang–Minlon Wolff–Kishner reduction conditions (hydrazine hydrate, potassium hydroxide, diethylene glycol, 195 °C) for the steroidal ketone shown above. The product was obtained in 79% yield compared to 91% obtained from the reduction via an intermediate N-tert-butyldimethylsilylhydrazone.
Side reactions
The Wolff–Kishner reduction is not suitable for base–sensitive substrates and can under certain conditions be hampered by steric hindrance surrounding the carbonyl group. Some of the more common side-reactions are listed below.
Azine formation
A commonly encountered side-reaction in Wolff–Kishner reductions involves azine formation by reaction of hydrazone with the carbonyl compound. Formation of the ketone can be suppressed by vigorous exclusion of water during the reaction. Several of the presented procedures require isolation of the hydrazone compound prior to reduction. This can be complicated by further transformation of the product hydrazone to the corresponding hydrazine during product purification. Cram found that azine formation is favored by rapid addition of preformed hydrazones to potassium tert-butoxide in anhydrous dimethylsulfoxide.[18]

Reduction of ketones to alcohols by sodium ethoxide
The second principal side reaction is the reduction of the ketone or aldehyde to the corresponding alcohol. After initial hydrolysis of the hydrazone, the free carbonyl derivative is reduced by alkoxide to the carbinol. In 1924, Eisenlohr reported that substantial amounts of hydroxydecalin were observed during the attempted Wolff–Kishner reduction of trans-β-decalone.[33] In general, alcohol formation may be repressed by exclusion of water or by addition of excess hydrazine.
Kishner–Leonard elimination
Kishner noted during his initial investigations that in some instances, α-substitution of a carbonyl group can lead to elimination affording unsaturated hydrocarbons under typical reaction conditions. Leonard later further developed this reaction and investigated the influence of different α-substituents on the reaction outcome.[23][34] He found that the amount of elimination increases with increasing steric bulk of the leaving group. Furthermore, α-dialkylamino-substituted ketones generally gave a mixture of reduction and elimination product whereas less basic leaving groups resulted in exclusive formation of the alkene product.

The fragmentation of α,β-epoxy ketones to allylic alcohols has been extended to a synthetically useful process and is known as the Wharton reaction.[35]
Cleavage or rearrangement of strained rings adjacent to the carbonyl group
Grob rearrangement of strained rings adjacent to the carbonyl group has been observed by Erman and coworkers.[36] During an attempted Wolff–Kishner reduction of trans-π-bromocamphor under Cram’s conditions, limonene was isolated as the only product.
Similarly, cleavage of strained rings adjacent to the carbonyl group can occur. When 9β,19-cyclo-5α-pregnane-3,11,20-trione 3,20-diethylene ketal was subjected to Huang–Minlon conditions, ring-enlargement was observed instead of formation of the 11-deoxo-compound.[37]

Applications
The Wolff–Kishner reduction is an effective tool in organic synthesis. For example, Ishibashi and coworkers employed the Huang Minlon modification of the Wolff–Kishner reduction as one of the final steps in their synthesis of (±)-aspidospermidine. Distillable material was removed after hydrazone formation at 160 °C and then heated to 210 °C overnight. The carbonyl group that was reduced in the Wolff–Kishner reduction was essential for preceding steps in the synthesis. The tertiary amide was stable to the reaction conditions and reduced subsequently by lithium aluminum hydride.[5]

Amides are usually not suitable substrates for the Wolff–Kishner reduction as demonstrated by the example above. Coe and coworkers found however that a twisted amide can be efficiently reduced under Wolff–Kishner conditions.[38] The authors explain this observation with the stereoelectronic bias of the substrate which prevents “anti–Bredt” iminium ion formation and therefore favors ejection of alcohol and hydrazone formation. The amide functionality in this strained substrate can be considered as isolated amine and ketone functionalities as resonance stabilization is prevented due to torsional restrictions. The product was obtained in 68% overall yield in a two step procedure.

In 2011, Pettus and Green reduced a tricyclic carbonyl compound using the Huang Minlon modification of the Wolff–Kishner reduction.[39] Several attempts towards decarbonylation of tricyclic allylic acetate containing ketone failed and the acetate functionality had to be removed to allow successful Wolff–Kishner reduction. Finally, the allylic alcohol was installed via oxyplumbation.

The Wolff–Kishner reduction has also been used on kilogram scale for the synthesis of a functionalized imidazole substrate. Several alternative reduction methods were investigated, but all of the tested conditions remained unsuccessful. Safety concerns for a large scale Wolff–Kishner reduction were addressed and a highly optimized procedure afforded to product in good yield.[40]

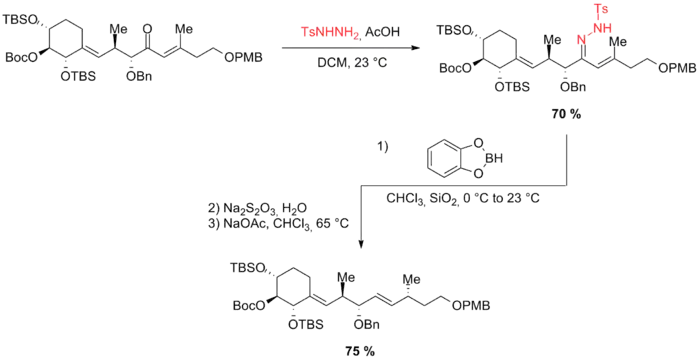
McIntosh et al. used an allylic diazene rearrangement in their synthesis of the C21–C34 fragment of antascomicin B.[41] The hydrazone was reduced selectively with catecholborane and excess reducing agent decomposed with sodium thiosulfate. The crude reaction product was then treated with sodium acetate and heated under reflux in chloroform to give the 1,4-syn isomer.
References
- Kishner, N (1911). "Wolff–Kishner reduction; Huang–Minlon modification". J. Russ. Phys. Chem. Soc. 43: 582–595.
- Wolff, L. (1912). "Chemischen Institut der Universität Jena: Methode zum Ersatz des Sauerstoffatoms der Ketone und Aldehyde durch Wasserstoff. [Erste Abhandlung.]". Justus Liebig's Annalen der Chemie. 394: 86–108. doi:10.1002/jlac.19123940107.
- Overman, L. E.; Ricca, D. J.; Tran, V. D. (1993). "First total synthesis of scopadulcic acid B". Journal of the American Chemical Society. 115 (5): 2042. doi:10.1021/ja00058a064.
- Marino, J. P.; Rubio, M. B.; Cao, G.; De Dios, A. (2002). "Total Synthesis of (+)-Aspidospermidine: A New Strategy for the Enantiospecific Synthesis of Aspidosperma Alkaloids". Journal of the American Chemical Society. 124 (45): 13398–13399. doi:10.1021/ja026357f. PMID 12418888.
- Kawano, M.; Kiuchi, T.; Negishi, S.; Tanaka, H.; Hoshikawa, T.; Matsuo, J. I.; Ishibashi, H. (2013). "Regioselective Inter- and Intramolecular Formal \4+2] Cycloaddition of Cyclobutanones with Indoles and Total Synthesis of (±)-Aspidospermidine". Angewandte Chemie International Edition. 52 (3): 906–10. doi:10.1002/anie.201206734. PMID 23184896.
- Miyaoka, H.; Kajiwara, Y.; Hara, Y.; Yamada, Y. (2001). "Total Synthesis of Natural Dysidiolide". The Journal of Organic Chemistry. 66 (4): 1429–1435. doi:10.1021/jo0015772. PMID 11312976.
- Lewis, D. E. (2013). "Disability, Despotism, Deoxygenation-From Exile to Academy Member: Nikolai Matveevich Kizhner". Angewandte Chemie International Edition. 52 (45): 11704–11712. doi:10.1002/anie.201303165. PMID 24123691.
- Herr, C. H.; Whitmore, F. C.; Schiessler, R. W. (1945). "The Wolff-Kishner Reaction at Atmospheric Pressure". Journal of the American Chemical Society. 67 (12): 2061. doi:10.1021/ja01228a002.
- Soffer, M. D.; Soffer, M. B.; Sherk, K. W. (1945). "A Low Pressure Method for Wolff—Kishner Reduction". Journal of the American Chemical Society. 67 (9): 1435. doi:10.1021/ja01225a004.
- Szmant, H. H.; Harmuth, C. M. (1964). "The Wolff-Kishner Reaction of Hydrazones". Journal of the American Chemical Society. 86 (14): 2909. doi:10.1021/ja01068a028.
- Szmant, H. H. (1968). "The Mechanism of the Wolff-Kishner Reduction, Elimination, and Isomerization Reactions". Angewandte Chemie International Edition in English. 7 (2): 120–128. doi:10.1002/anie.196801201.
- Szmant, H. H.; Roman, M. N. (1966). "The Effect of Dimethyl Sulfoxide on the Rate of the Wolff-Kishner Reaction of Benzophenone Hydrazone1". Journal of the American Chemical Society. 88 (17): 4034. doi:10.1021/ja00969a025.
- Szmant, H. H.; Alciaturi, C. E. (1977). "Mechanistic aspects of the Wolff-Kishner reaction. 6. Comparison of the hydrazones of benzophenone, fluorenone, dibenzotropone, and dibenzosuberone". The Journal of Organic Chemistry. 42 (6): 1081. doi:10.1021/jo00426a034.
- Szmant, H. H.; Harnsberger, H. F.; Butler, T. J.; Barie, W. P. (1952). "Kinetics of the Wolff-Kishner Reaction of Diaryl Ketone Hydrazones". Journal of the American Chemical Society. 74 (11): 2724. doi:10.1021/ja01131a009.
- Taber, D. F.; Stachel, S. J. (1992). "On the mechanism of the Wolff-Kishner reduction". Tetrahedron Letters. 33 (7): 903. doi:10.1016/S0040-4039(00)91571-5.
- Huang-Minlon, [N. A. (1946). "A Simple Modification of the Wolff-Kishner Reduction". Journal of the American Chemical Society. 68 (12): 2487–2488. doi:10.1021/ja01216a013.
- Osdene, T. S.; Timmis, G. M.; Maguire, M. H.; Shaw, G.; Goldwhite, H.; Saunders, B. C.; Clark, E. R.; Epstein, P. F.; Lamchen, M.; Stephen, A. M.; Tipper, C. F. H.; Eaborn, C.; Mukerjee, S. K.; Seshadri, T. R.; Willenz, J.; Robinson, R.; Thomas, A. F.; Hickman, J. R.; Kenyon, J.; Crocker, H. P.; Hall, R. H.; Burnell, R. H.; Taylor, W. I.; Watkins, W. M.; Barton, D. H. R.; Ives, D. A. J.; Thomas, B. R. (1955). "Notes". Journal of the Chemical Society (Resumed): 2038. doi:10.1039/JR9550002038.
- Cram, D. J.; Sahyun, M. R. V. (1962). "Room Temperature Wolff-Kishner Reduction and Cope Elimination Reactions". Journal of the American Chemical Society. 84 (9): 1734. doi:10.1021/ja00868a048.
- Grundon, M. F.; Henbest, H. B.; Scott, M. D. (1963). "344. The reactions of hydrazones and related compounds with strong bases. Part I. A modified Wolff?Kishner procedure". Journal of the Chemical Society (Resumed): 1855–1858. doi:10.1039/JR9630001855.
- Caglioti, L.; Magi, M. (1963). "The reaction of tosylhydrazones with lithium aluminium hydride". Tetrahedron. 19 (7): 1127. doi:10.1016/S0040-4020(01)98571-0.
- Furrow, M. E.; Myers, A. G. (2004). "Practical Procedures for the Preparation ofN-tert-Butyldimethylsilylhydrazones and Their Use in Modified Wolff−Kishner Reductions and in the Synthesis of Vinyl Halides andgem-Dihalides". Journal of the American Chemical Society. 126 (17): 5436–5445. doi:10.1021/ja049694s. PMID 15113215.
- Huang-Minlon, [N. A. . (1949). "Reduction of Steroid Ketones and other Carbonyl Compounds by Modified Wolff--Kishner Method". Journal of the American Chemical Society. 71 (10): 3301–3303. doi:10.1021/ja01178a008.
- Leonard, N. J.; Gelfand, S. (1955). "The Kishner Reduction-Elimination. II. α-Substituted Pinacolones1,2". Journal of the American Chemical Society. 77 (12): 3272. doi:10.1021/ja01617a036.
- Caglioti, L. (1966). "The reduction of tosylhydrazones and of acyl tosylhydrazides". Tetrahedron. 22 (2): 487–493. doi:10.1016/0040-4020(66)80015-7.
- Hutchins, R. O.; Milewski, C. A.; Maryanoff, B. E. (1973). "Selective deoxygenation of ketones and aldehydes including hindered systems with sodium cyanoborohydride". Journal of the American Chemical Society. 95 (11): 3662. doi:10.1021/ja00792a033.
- Hutchins, R. O. (1991). Comp. Org. Synth. Pergamon. pp. 327–362.
- Miller, V. P.; Yang, D. Y.; Weigel, T. M.; Han, O.; Liu, H. W. (1989). "Studies of the mechanistic diversity of sodium cyanoborohydride reduction of tosylhydrazones". The Journal of Organic Chemistry. 54 (17): 4175. doi:10.1021/jo00278a035.
- Bosch, J.; Bonjoch, J. (1981). "Synthetic route to 6-functionalized 2-azabicyclo\3.3.1]nonanes". The Journal of Organic Chemistry. 46 (8): 1538. doi:10.1021/jo00321a004.
- Hutchins, R. O.; Kacher, M.; Rua, L. (1975). "Synthetic utility and mechanism of the reductive deoxygenation of .alpha.,.beta.-unsaturated p-tosylhydrazones with sodium cyanoborohydride". The Journal of Organic Chemistry. 40 (7): 923. doi:10.1021/jo00895a024.
- Kabalka, G. W.; Yang, D. T. C.; Baker, J. D. (1976). "Deoxygenation of .alpha.,.beta.-unsaturated aldehydes and ketones via the catecholborane reduction of the corresponding tosylhydrazones". The Journal of Organic Chemistry. 41 (3): 574. doi:10.1021/jo00865a043.
- Taylor, E. J.; Djerassi, C. (1976). "Mechanism of the sodium cyanoborohydride reduction of .alpha.,.beta.-unsaturated tosylhydrazones". Journal of the American Chemical Society. 98 (8): 2275. doi:10.1021/ja00424a046.
- Qi, W.; McIntosh, M. C. (2008). "Acyclic 1,4-Stereocontrol via Reductive 1,3-Transpositions". Organic Letters. 10 (2): 357–359. doi:10.1021/ol702921x. PMC 2613761. PMID 18092798.
- Eisenlohr, F.; Polenske, R. (1924). "Über die raumisomeren Formen des Dekahydro-naphthalins (Dekalins)". Berichte der Deutschen Chemischen Gesellschaft (A and B Series). 57 (9): 1639. doi:10.1002/cber.19240570902.
- Leonard, N. J.; Gelfand, S. (1955). "The Kishner Reduction-Elimination. I. Cyclic and Open Chain α-Aminoketones1,2". Journal of the American Chemical Society. 77 (12): 3269. doi:10.1021/ja01617a035.
- Wharton, P.; Bohlen, D. (1961). "Communications- Hydrazine Reduction of α, β-Epoxy Ketones to Allylic Alcohols". The Journal of Organic Chemistry. 26 (9): 3615. doi:10.1021/jo01067a117.
- Gustafson, D. H.; Erman, W. F. (1965). "A Novel Fragmentation of trans-π-Bromocamphor". The Journal of Organic Chemistry. 30 (5): 1665. doi:10.1021/jo01016a516.
- Kupchan, S. M.; Abushanab, E.; Shamasundar, K. T.; By, A. W. (1967). "Buxus alkaloids. 13. A synthetic approach to the 9(10--19) abeo-pregnane system". Journal of the American Chemical Society. 89 (24): 6327–6332. doi:10.1021/ja01000a060. PMID 6066048.
- Bashore, C. G.; Samardjiev, I. J.; Bordner, J.; Coe, J. W. (2003). "Twisted Amide Reduction under Wolff−Kishner Conditions: Synthesis of a Benzo-1-Aza-Adamantane Derivative". Journal of the American Chemical Society. 125 (11): 3268–3272. doi:10.1021/ja028152c. PMID 12630882.
- Green, J. C.; Pettus, T. R. R. (2011). "An Oxidative Dearomatization-Induced \5 + 2] Cascade Enabling the Syntheses of α-Cedrene, α-Pipitzol, andsec-Cedrenol". Journal of the American Chemical Society. 133 (5): 1603–1608. doi:10.1021/ja109925g. PMID 21194216.
- Kuethe, J. T.; Childers, K. G.; Peng, Z.; Journet, M.; Humphrey, G. R.; Vickery, T.; Bachert, D.; Lam, T. T. (2009). "A Practical, Kilogram-Scale Implementation of the Wolff−Kishner Reduction". Organic Process Research & Development. 13 (3): 576. doi:10.1021/op9000274.
- Hutchison, John M.; Gibson, Andrew S.; Williams, David T.; McIntosh, Matthias C. (2011). "Synthesis of the C21–C34 fragment of antascomicin B". Tetrahedron Letters. 52 (48): 6349–6351. doi:10.1016/j.tetlet.2011.09.027. ISSN 0040-4039. PMC 3244276. PMID 22199407.
Further reading
- Todd, D. The Wolff-Kishner Reduction. In Org. React. (eds. Adams, E.); John-Wiley & Sons, Inc.: London, 1948, 4, 378
- Hutchins, R. O. Reduction of C=X to CH2 by Wolff-Kishner and Other Hydrazone Methods. In Comp. Org. Synth. (eds. Trost, B. M., Fleming, I.); Pergamon: Oxford, 1991, 8, 327
- Lewis, D. E. The Wolff-Kishner Reduction and Related Reactions. Discovery and Development; Elsevier: Amsterdam, 2019. ISBN 9780128157275.