Argininosuccinate lyase
ASL (argininosuccinate lyase, also known as argininosuccinase) is an enzyme that catalyzes the reversible breakdown of argininosuccinate (ASA) producing the amino acid arginine and dicarboxylic acid fumarate. Located in liver cytosol, ASL is the fourth enzyme of the urea cycle and involved in the biosynthesis of arginine in all species and the production of urea in ureotelic species.[2] Mutations in ASL, resulting low activity of the enzyme, increase levels of urea in the body and result in various side effects.
| Argininosuccinate lyase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Crystal structure of duck argininosuccinate lyase with bound argininosuccinate.[1] | |||||||||
| Identifiers | |||||||||
| EC number | 4.3.2.1 | ||||||||
| CAS number | 9027-34-3 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
| Argininosuccinate lyase | |||||||
|---|---|---|---|---|---|---|---|
 Crystallographic structure of the human ASL monomer with labeled domains.[2] | |||||||
| Identifiers | |||||||
| Symbol | ASL | ||||||
| NCBI gene | 435 | ||||||
| HGNC | 746 | ||||||
| OMIM | 608310 | ||||||
| RefSeq | NM_000048 | ||||||
| UniProt | P04424 | ||||||
| Other data | |||||||
| EC number | 4.3.2.1 | ||||||
| Locus | Chr. 7 pter-q22 | ||||||
| |||||||
The ASL gene is located on chromosome 7 between the centromere (junction of the long and short arm) and the long (q) arm at position 11.2, from base pair 64,984,963 to base pair 65,002,090.
ASL is related to intragenic complementation.[3][4][5]
Structure
ASL is composed of four identical monomers; each monomer consisting of a single polypeptide chain between 49 and 52 kDa,[6] between 196 and 208 kDa for the entire tetrameric enzyme. Each monomer has three highly conserved regions remote from one another, but these regions cluster together in the tetramer to form four active sites. Therefore, each ASL homotetramer has four active sites to catalyze the breakdown of argininosuccinate.
Each monomer in the ASL homotetramer is composed of three structural domains; all three are primarily alpha helical. Domains 1 and 3 are similar in structure as they both consist of helix-turn-helix motifs. Domain 1 of the monomer contains the amino terminus. Domain 2 contains one small beta sheet, nine alpha helices, and the carboxyl terminus. Three of the nine alpha helices on one monomer are engaged mainly in hydrophobic interactions with another monomer to form a dimer. Two dimers then associate by way of alpha helix, one from each monomer, to form a central 20-helix core. The association of all four monomers allows for the catalytic activity at each possible active site.[4]
Intragenic complementation
Multiple copies of a polypeptide encoded by a gene often can form an aggregate referred to as a multimer. When a multimer is formed from polypeptides produced by two different mutant alleles of a particular gene, the mixed multimer may exhibit greater functional activity than the unmixed multimers formed by each of the mutants alone. When a mixed multimer displays increased functionality relative to the unmixed multimers, the phenomenon is referred to as intragenic complementation. In humans, ASL is a multimer (tetramer) protein. An ASL disorder in humans can arise from mutations in the ASL gene, particularly mutations that affect the active site of the mutant multimer protein. ASL disorder is associated with considerable clinical and genetic heterogeneity which is considered to reflect the extensive intragenic complementation occurring among individual patients.[3][4][5]
Mechanism
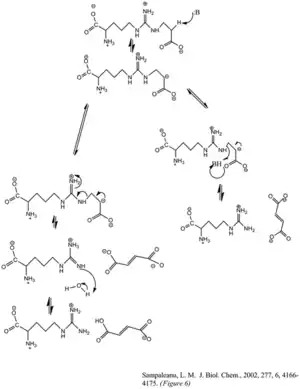

The enzyme's cleavage of the argininosuccinate, to form fumarate and arginine, occurs through an E1cb elimination reaction. The base initiates the reaction by deprotonating the carbon adjacent to the arginine, or leaving group. Recent mutagenic studies of ASL homologues have shown that Histidine 162 or Threonine 161 of ASL is responsible for the proton abstraction of the Cβ, either directly or indirectly through a water molecule.[6] Lysine 289 is thought to stabilize the negatively charged carbanion intermediate. Although there is no consensus of the catalytic acid that donates the proton to the imine functional group of the arginine product, some mutagenesis studies show serine 283 may be involved.[6]
Role in the urea cycle
Ammonia (NH3) is a toxic substance for many aerobic organisms and must be excreted. Some aquatic organisms release the toxin right directly into their environment, while other ureotelic species must convert their toxic nitrogen waste into non-toxic components, like uric acid or urea, through a series of catalyzed steps better known as the urea cycle. ASL catalyzes the fourth step in the cycle, following the action of argininosuccinate synthetase (ASS) in the liver cytosol. While ASS catalyzes the formation of argininosuccinate from citrulline and aspartate, ASL breaks the newly formed argininosuccinate into L-arginine and fumarate. L-arginine continues through the urea cycle to form urea and ornithine, while fumarate can enter the citric acid cycle.[7]
δ-Crystallin
ASL, δ-crystallin, class II fumarase, aspartase, adenylosuccinase lyase, and 3-carboxy-cis and cis-muconate lactonizing enzyme are all members of the same homotetrameric superfamily of enzymes, in which most catalyze the same type of elimination reactions where a C-O or C-N bond is broken and fumarate is released as a product. δ-crystallins are the major structural eye lens water-soluble proteins of most birds, reptiles, and some other vertebrates.[4]
Within the superfamily, ASL is most closely related to δ-crystallin in amino acid sequence and in protein fold structure. There are two isoforms of δ-crystallin, δI and δII. These two isoforms conserve 69% and 71% of the ASL amino acid sequence, respectively, but only the δII isoform retains the same enzymatic activity as ASL. The similarities have led researches to believe that these crystallins have evolved from the recruitment to the lens of preexisting metabolic enzymes, like ASL, by a process called 'gene sharing'. The same gene product functions as both a lens crystallin and an enzyme in other non-ocular tissues. Comparative studies of the δ-crystallins have been beneficial for understanding the enzymatic mechanism of the ASL reaction.[8]
Mutations and ASL deficiencies: argininosuccinic aciduria
Mutations in the human ASL gene causes argininosuccinic aciduria, a rare autosomal recessive disorder, and results in deficiencies of the urea cycle. Argininosuccinate lyase is an intermediate enzyme in the urea synthesis pathway and its function is imperative to the continuation of the cycle. A non-functioning enzyme results in patients' accumulation of ammonia, argininosuccinate, and citrulline in the blood, and argininosuccinate is excreted in the urine.[9] Other resulting symptoms include lethargy, vomiting, hypothermia, hyperventilation, hepatomegaly and progressive encephalopathy in infant patients, and abnormal hair growth, hepatic fibrosis, episodic vomiting, growth and developmental delay,[9] in patients experiencing the disorder later in childhood.
ASL is a key enzyme in the conversion of ammonia to urea through the urea cycle. Ammonia builds to toxic levels, resulting in hyperammonemia.[10] Ammonia is toxic in part because it affects the nervous system. There is biochemical evidence that shows rises in ammonia can inhibit glutaminase and therefore limit the rate of synthesis of neurotransmitters such as glutamate,[11] which can explain the developmental delay in argininosuccinic aciduria patients.
One mutation in patients with argininosuccinic aciduria occurs when glutamine 286 is mutated to arginine. The enzyme now has a positively charged arginine in place of a neutrally charged glutamine and studies suggest this change may sterically and/or electrostatically hinder a conformational change necessary for catalysis.
References
- PDB: 1TJW; Sampaleanu LM, Codding PW, Lobsanov YD, Tsai M, Smith GD, Horvatin C, Howell PL (December 2004). "Structural studies of duck delta2 crystallin mutants provide insight into the role of Thr161 and the 280s loop in catalysis". Biochem. J. 384 (Pt 2): 437–47. doi:10.1042/BJ20040656. PMC 1134128. PMID 15320872.
- PDB: 1K62; Sampaleanu LM, Vallée F, Thompson GD, Howell PL (December 2001). "Three-dimensional structure of the argininosuccinate lyase frequently complementing allele Q286R". Biochemistry. 40 (51): 15570–80. doi:10.1021/bi011525m. PMID 11747432.
- Turner MA, Simpson A, McInnes RR, Howell PL (August 1997). "Human argininosuccinate lyase: a structural basis for intragenic complementation". Proc. Natl. Acad. Sci. U.S.A. 94 (17): 9063–8. doi:10.1073/pnas.94.17.9063. PMC 23030. PMID 9256435.
- Yu B, Howell PL (October 2000). "Intragenic complementation and the structure and function of argininosuccinate lyase". Cell. Mol. Life Sci. 57 (11): 1637–51. doi:10.1007/PL00000646. PMID 11092456. S2CID 1254964.
- Yu B, Thompson GD, Yip P, Howell PL, Davidson AR (December 2001). "Mechanisms for intragenic complementation at the human argininosuccinate lyase locus". Biochemistry. 40 (51): 15581–90. doi:10.1021/bi011526e. PMID 11747433.
- Sampaleanu LM, Yu B, Howell PL (February 2002). "Mutational analysis of duck delta 2 crystallin and the structure of an inactive mutant with bound substrate provide insight into the enzymatic mechanism of argininosuccinate lyase". J. Biol. Chem. 277 (6): 4166–75. doi:10.1074/jbc.M107465200. PMID 11698398.
- Pratt, Charlotte Amerley; Voet, Donald; Voet, Judith G. (2008). "Figure 20.8". Fundamentals of biochemistry: life at the molecular level. New York: Wiley. ISBN 978-0-470-12930-2.
- Chakraborty AR, Davidson A, Howell PL (February 1999). "Mutational analysis of amino acid residues involved in argininosuccinate lyase activity in duck delta II crystallin". Biochemistry. 38 (8): 2435–43. doi:10.1021/bi982150g. PMID 10029537.
- Ficicioglu C, Mandell R, Shih VE (November 2009). "Argininosuccinate lyase deficiency: longterm outcome of 13 patients detected by newborn screening". Mol. Genet. Metab. 98 (3): 273–7. doi:10.1016/j.ymgme.2009.06.011. PMC 2773214. PMID 19635676.
- "ASL gene argininosuccinate lyase". NIH. U.S. Department of Health & Human Services. 2007.
- Jack, JJB (1982). "Actions of ammonia on the central nervous system". Journal of Inherited Metabolic Disease. 5 (S2): 104. doi:10.1007/BF01805572. S2CID 33915515.