Calorimetry
Calorimetry is the science or act of measuring changes in state variables of a body for the purpose of deriving the heat transfer associated with changes of its state due, for example, to chemical reactions, physical changes, or phase transitions under specified constraints. Calorimetry is performed with a calorimeter. The word calorimetry is derived from the Latin word calor, meaning heat and the Greek word μέτρον (metron), meaning measure. Scottish physician and scientist Joseph Black, who was the first to recognize the distinction between heat and temperature, is said to be the founder of the science of calorimetry.[2]


Indirect calorimetry calculates heat that living organisms produce by measuring either their production of carbon dioxide and nitrogen waste (frequently ammonia in aquatic organisms, or urea in terrestrial ones), or from their consumption of oxygen. Lavoisier noted in 1780 that heat production can be predicted from oxygen consumption this way, using multiple regression. The dynamic energy budget theory explains why this procedure is correct. Heat generated by living organisms may also be measured by direct calorimetry, in which the entire organism is placed inside the calorimeter for the measurement.
A widely used modern instrument is the differential scanning calorimeter, a device which allows thermal data to be obtained on small amounts of material. It involves heating the sample at a controlled rate and recording the heat flow either into or from the specimen.
Classical calorimetric calculation of heat
Basic classical calculation with respect to volume
Calorimetry requires that a reference material that changes temperature have known definite thermal constitutive properties. The classical rule, recognized by Clausius and Kelvin, is that the pressure exerted by the calorimetric material is fully and rapidly determined solely by its temperature and volume; this rule is for changes that do not involve phase change, such as melting of ice. There are many materials that do not comply with this rule, and for them, the present formula of classical calorimetry does not provide an adequate account. Here the classical rule is assumed to hold for the calorimetric material being used, and the propositions are mathematically written:
The thermal response of the calorimetric material is fully described by its pressure as the value of its constitutive function of just the volume and the temperature . All increments are here required to be very small. This calculation refers to a domain of volume and temperature of the body in which no phase change occurs, and there is only one phase present. An important assumption here is continuity of property relations. A different analysis is needed for phase change




When a small increment of heat is gained by a calorimetric body, with small increments, of its volume, and of its temperature, the increment of heat, , gained by the body of calorimetric material, is given by




where
- denotes the latent heat with respect to volume, of the calorimetric material at constant controlled temperature . The surroundings' pressure on the material is instrumentally adjusted to impose a chosen volume change, with initial volume . To determine this latent heat, the volume change is effectively the independently instrumentally varied quantity. This latent heat is not one of the widely used ones, but is of theoretical or conceptual interest.
- denotes the heat capacity, of the calorimetric material at fixed constant volume , while the pressure of the material is allowed to vary freely, with initial temperature . The temperature is forced to change by exposure to a suitable heat bath. It is customary to write simply as , or even more briefly as . This latent heat is one of the two widely used ones.[3][4][5][6][7][8][9]





The latent heat with respect to volume is the heat required for unit increment in volume at constant temperature. It can be said to be 'measured along an isotherm', and the pressure the material exerts is allowed to vary freely, according to its constitutive law . For a given material, it can have a positive or negative sign or exceptionally it can be zero, and this can depend on the temperature, as it does for water about 4 C.[10][11][12][13] The concept of latent heat with respect to volume was perhaps first recognized by Joseph Black in 1762.[14] The term 'latent heat of expansion' is also used.[15] The latent heat with respect to volume can also be called the 'latent energy with respect to volume'. For all of these usages of 'latent heat', a more systematic terminology uses 'latent heat capacity'.

The heat capacity at constant volume is the heat required for unit increment in temperature at constant volume. It can be said to be 'measured along an isochor', and again, the pressure the material exerts is allowed to vary freely. It always has a positive sign. This means that for an increase in the temperature of a body without change of its volume, heat must be supplied to it. This is consistent with common experience.
Quantities like are sometimes called 'curve differentials', because they are measured along curves in the surface.

Classical theory for constant-volume (isochoric) calorimetry
Constant-volume calorimetry is calorimetry performed at a constant volume. This involves the use of a constant-volume calorimeter. Heat is still measured by the above-stated principle of calorimetry.
This means that in a suitably constructed calorimeter, called a bomb calorimeter, the increment of volume can be made to vanish, . For constant-volume calorimetry:


where
- denotes the increment in temperature and
- denotes the heat capacity at constant volume.
Classical heat calculation with respect to pressure
From the above rule of calculation of heat with respect to volume, there follows one with respect to pressure.[3][7][16][17]
In a process of small increments, of its pressure, and of its temperature, the increment of heat, , gained by the body of calorimetric material, is given by


where
- denotes the latent heat with respect to pressure, of the calorimetric material at constant temperature, while the volume and pressure of the body are allowed to vary freely, at pressure and temperature ;
- denotes the heat capacity, of the calorimetric material at constant pressure, while the temperature and volume of the body are allowed to vary freely, at pressure and temperature . It is customary to write simply as , or even more briefly as .




The new quantities here are related to the previous ones:[3][7][17][18]


where
- denotes the partial derivative of with respect to evaluated for

and
- denotes the partial derivative of with respect to evaluated for .

The latent heats and are always of opposite sign.[19]
It is common to refer to the ratio of specific heats as


Calorimetry through phase change, equation of state shows one jump discontinuity
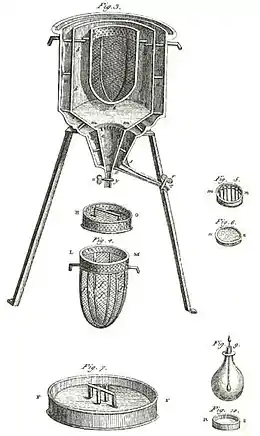
An early calorimeter was that used by Laplace and Lavoisier, as shown in the figure above. It worked at constant temperature, and at atmospheric pressure. The latent heat involved was then not a latent heat with respect to volume or with respect to pressure, as in the above account for calorimetry without phase change. The latent heat involved in this calorimeter was with respect to phase change, naturally occurring at constant temperature. This kind of calorimeter worked by measurement of mass of water produced by the melting of ice, which is a phase change.
Cumulation of heating
For a time-dependent process of heating of the calorimetric material, defined by a continuous joint progression of and , starting at time and ending at time , there can be calculated an accumulated quantity of heat delivered, . This calculation is done by mathematical integration along the progression with respect to time. This is because increments of heat are 'additive'; but this does not mean that heat is a conservative quantity. The idea that heat was a conservative quantity was invented by Lavoisier, and is called the 'caloric theory'; by the middle of the nineteenth century it was recognized as mistaken. Written with the symbol , the quantity is not at all restricted to be an increment with very small values; this is in contrast with .







One can write


- .

This expression uses quantities such as which are defined in the section below headed 'Mathematical aspects of the above rules'.

Mathematical aspects of the above rules
The use of 'very small' quantities such as is related to the physical requirement for the quantity to be 'rapidly determined' by and ; such 'rapid determination' refers to a physical process. These 'very small' quantities are used in the Leibniz approach to the infinitesimal calculus. The Newton approach uses instead 'fluxions' such as , which makes it more obvious that must be 'rapidly determined'.

In terms of fluxions, the above first rule of calculation can be written[22]

where
- denotes the time

- denotes the time rate of heating of the calorimetric material at time
- denotes the time rate of change of volume of the calorimetric material at time

- denotes the time rate of change of temperature of the calorimetric material.

The increment and the fluxion are obtained for a particular time that determines the values of the quantities on the righthand sides of the above rules. But this is not a reason to expect that there should exist a mathematical function . For this reason, the increment is said to be an 'imperfect differential' or an 'inexact differential'.[23][24][25] Some books indicate this by writing instead of .[26][27] Also, the notation đQ is used in some books.[23][28] Carelessness about this can lead to error.[29]


The quantity is properly said to be a functional of the continuous joint progression of and , but, in the mathematical definition of a function, is not a function of . Although the fluxion is defined here as a function of time , the symbols and respectively standing alone are not defined here.

Physical scope of the above rules of calorimetry
The above rules refer only to suitable calorimetric materials. The terms 'rapidly' and 'very small' call for empirical physical checking of the domain of validity of the above rules.
The above rules for the calculation of heat belong to pure calorimetry. They make no reference to thermodynamics, and were mostly understood before the advent of thermodynamics. They are the basis of the 'thermo' contribution to thermodynamics. The 'dynamics' contribution is based on the idea of work, which is not used in the above rules of calculation.
Experimentally conveniently measured coefficients
Empirically, it is convenient to measure properties of calorimetric materials under experimentally controlled conditions.
Pressure increase at constant volume
For measurements at experimentally controlled volume, one can use the assumption, stated above, that the pressure of the body of calorimetric material is can be expressed as a function of its volume and temperature.
For measurement at constant experimentally controlled volume, the isochoric coefficient of pressure rise with temperature, is defined by
- .[30]

Expansion at constant pressure
For measurements at experimentally controlled pressure, it is assumed that the volume of the body of calorimetric material can be expressed as a function of its temperature and pressure . This assumption is related to, but is not the same as, the above used assumption that the pressure of the body of calorimetric material is known as a function of its volume and temperature; anomalous behaviour of materials can affect this relation.

The quantity that is conveniently measured at constant experimentally controlled pressure, the isobaric volume expansion coefficient, is defined by

Compressibility at constant temperature
For measurements at experimentally controlled temperature, it is again assumed that the volume of the body of calorimetric material can be expressed as a function of its temperature and pressure , with the same provisos as mentioned just above.
The quantity that is conveniently measured at constant experimentally controlled temperature, the isothermal compressibility, is defined by

Relation between classical calorimetric quantities
Assuming that the rule is known, one can derive the function of that is used above in the classical heat calculation with respect to pressure. This function can be found experimentally from the coefficients and through the mathematically deducible relation



- .[37]

Connection between calorimetry and thermodynamics
Thermodynamics developed gradually over the first half of the nineteenth century, building on the above theory of calorimetry which had been worked out before it, and on other discoveries. According to Gislason and Craig (2005): "Most thermodynamic data come from calorimetry..."[38] According to Kondepudi (2008): "Calorimetry is widely used in present day laboratories."[39]
In terms of thermodynamics, the internal energy of the calorimetric material can be considered as the value of a function of , with partial derivatives and .




Then it can be shown that one can write a thermodynamic version of the above calorimetric rules:

with

and

Again, further in terms of thermodynamics, the internal energy of the calorimetric material can sometimes, depending on the calorimetric material, be considered as the value of a function of , with partial derivatives and , and with being expressible as the value of a function of , with partial derivatives and .






Then, according to Adkins (1975),[44] it can be shown that one can write a further thermodynamic version of the above calorimetric rules:

with

and
- .[44]

Beyond the calorimetric fact noted above that the latent heats and are always of opposite sign, it may be shown, using the thermodynamic concept of work, that also

Special interest of thermodynamics in calorimetry: the isothermal segments of a Carnot cycle
Calorimetry has a special benefit for thermodynamics. It tells about the heat absorbed or emitted in the isothermal segment of a Carnot cycle.
A Carnot cycle is a special kind of cyclic process affecting a body composed of material suitable for use in a heat engine. Such a material is of the kind considered in calorimetry, as noted above, that exerts a pressure that is very rapidly determined just by temperature and volume. Such a body is said to change reversibly. A Carnot cycle consists of four successive stages or segments:
(1) a change in volume from a volume to a volume at constant temperature so as to incur a flow of heat into the body (known as an isothermal change)



(2) a change in volume from to a volume at a variable temperature just such as to incur no flow of heat (known as an adiabatic change)

(3) another isothermal change in volume from to a volume at constant temperature such as to incur a flow or heat out of the body and just such as to precisely prepare for the following change


(4) another adiabatic change of volume from back to just such as to return the body to its starting temperature .
In isothermal segment (1), the heat that flows into the body is given by

and in isothermal segment (3) the heat that flows out of the body is given by
- .[46]

Because the segments (2) and (4) are adiabats, no heat flows into or out of the body during them, and consequently the net heat supplied to the body during the cycle is given by
- .

This quantity is used by thermodynamics and is related in a special way to the net work done by the body during the Carnot cycle. The net change of the body's internal energy during the Carnot cycle, , is equal to zero, because the material of the working body has the special properties noted above.

Special interest of calorimetry in thermodynamics: relations between classical calorimetric quantities
Relation of latent heat with respect to volume, and the equation of state
The quantity , the latent heat with respect to volume, belongs to classical calorimetry. It accounts for the occurrence of energy transfer by work in a process in which heat is also transferred; the quantity, however, was considered before the relation between heat and work transfers was clarified by the invention of thermodynamics. In the light of thermodynamics, the classical calorimetric quantity is revealed as being tightly linked to the calorimetric material's equation of state . Provided that the temperature is measured in the thermodynamic absolute scale, the relation is expressed in the formula

- .[47]



Practical constant-volume calorimetry (bomb calorimetry) for thermodynamic studies
Constant-volume calorimetry is calorimetry performed at a constant volume. This involves the use of a constant-volume calorimeter.
No work is performed in constant-volume calorimetry, so the heat measured equals the change in internal energy of the system. The heat capacity at constant volume is assumed to be independent of temperature.
Heat is measured by the principle of calorimetry.

where
- ΔU is change in internal energy,
- ΔT is change in temperature and
- CV is the heat capacity at constant volume.
In constant-volume calorimetry the pressure is not held constant. If there is a pressure difference between initial and final states, the heat measured needs adjustment to provide the enthalpy change. One then has

where
- ΔH is change in enthalpy and
- V is the unchanging volume of the sample chamber.
See also
- Isothermal microcalorimetry (IMC)
- Isothermal titration calorimetry
- Sorption calorimetry
- Reaction calorimeter
References
- Reardon, Francis D.; Leppik, Kalle E.; Wegmann, René; Webb, Paul; Ducharme, Miche B.; & Kenny, Glen P. (2006). The Snellen human calorimeter revisited, re-engineered and upgraded: design and performance characteristics. Med Bio Eng Comput, 44:721–728.
- Laidler, Keith, J. (1993). The World of Physical Chemistry. Oxford University Press. ISBN 0-19-855919-4.
- Bryan, G.H. (1907), pages 21–22.
- Partington, J.R. (1949), pages 155–157.
- Prigogine, I., Defay, R. (1950/1954). Chemical Thermodynamics, Longmans, Green & Co, London, pages 22-23.
- Crawford, F.H. (1963), Section 5.9, pp. 120–121.
- Adkins, C.J. (1975), Section 3.6, pages 43-46.
- Truesdell, C., Bharatha, S. (1977), pages 20-21.
- Landsberg, P.T. (1978), page 11.
- Maxwell, J.C. (1872), pages 232-233.
- Lewis, G.N., Randall, M. (1923/1961), pages 378-379.
- Truesdell, C., Bharatha, S. (1977), pages 9-10, 15-18, 36-37.
- Truesdell, C.A. (1980). The Tragicomical History of Thermodynamics, 1822–1854, Springer, New York, ISBN 0-387-90403-4.
- Lewis, G.N., Randall, M. (1923/1961), page 29.
- Maxwell, J.C. (1872), page 73.
- Crawford, F.H. (1963), Section 5.10, pp. 121–122.
- Truesdell, C., Bharatha, S. (1977), page 23.
- Crawford, F.H. (1963), Section 5.11, pp. 123–124.
- Truesdell, C., Bharatha, S. (1977), page 24.
- Truesdell, C., Bharatha, S. (1977), page 25.
- Kondepudi, D. (2008), pages 66-67.
- Truesdell, C., Bharatha, S. (1977), page 20.
- Adkins, C.J. (1975), Section 1.9.3, page 16.
- Landsberg, P.T. (1978), pages 8-9.
- An account of this is given by Landsberg, P.T. (1978), Chapter 4, pages 26-33.
- Fowler, R., Guggenheim, E.A. (1939/1965). Statistical Thermodynamics. A version of Statistical Mechanics for Students of Physics and Chemistry, Cambridge University Press, Cambridge UK, page 57.
- Guggenheim, E.A. (1949/1967), Section 1.10, pages 9-11.
- Lebon, G., Jou, D., Casas-Vázquez, J. (2008). Understanding Non-equilibrium Thermodynamics: Foundations, Applications, Frontiers, Springer-Verlag, Berlin, ISBN 978-3-540-74252-4, page 7.
- Planck, M. (1923/1926), page 57.
- Iribarne, J.V., Godson, W.L. (1973/1981), page 46.
- Lewis, G.N., Randall, M. (1923/1961), page 54.
- Guggenheim, E.A. (1949/1967), page 38.
- Callen, H.B. (1960/1985), page 84.
- Adkins, C.J. (1975), page 38.
- Bailyn, M. (1994), page 49.
- Kondepudi, D. (2008), page 180.
- Kondepudi, D. (2008), page 181.
- Gislason, E.A., Craig, N.C. (2005). Cementing the foundations of thermodynamics:comparison of system-based and surroundings-based definitions of work and heat, J. Chem. Thermodynamics 37: 954-966.
- Kondepudi, D. (2008), page 63.
- Preston, T. (1894/1904). The Theory of Heat, second edition, revised by J.R. Cotter, Macmillan, London, pages 700-701.
- Adkins, C.J. (1975), page 45.
- Truesdell, C., Bharatha, S. (1977), page 134.
- Kondepudi, D. (2008), page 64.
- Adkins, C.J. (1975), page 46.
- Truesdell, C., Bharatha, S. (1977), page 59.
- Truesdell, C., Bharatha, S. (1977), pages 52-53.
- Truesdell, C., Bharatha, S. (1977), page 150.
- Callen, H.B. (1960/1985), page 86.
Books
- Adkins, C.J. (1975). Equilibrium Thermodynamics, second edition, McGraw-Hill, London, ISBN 0-07-084057-1.
- Bailyn, M. (1994). A Survey of Thermodynamics, American Institute of Physics, New York, ISBN 0-88318-797-3.
- Bryan, G.H. (1907). Thermodynamics. An Introductory Treatise dealing mainly with First Principles and their Direct Applications, B.G. Tuebner, Leipzig.
- Callen, H.B. (1960/1985). Thermodynamics and an Introduction to Thermostatistics, second edition, Wiley, New York, ISBN 981-253-185-8.
- Crawford, F.H. (1963). Heat, Thermodynamics, and Statistical Physics, Rupert Hart-Davis, London, Harcourt, Brace, & World.
- Guggenheim, E.A. (1949/1967). Thermodynamics. An Advanced Treatment for Chemists and Physicists, North-Holland, Amsterdam.
- Iribarne, J.V., Godson, W.L. (1973/1981), Atmospheric Thermodynamics, second edition, D. Reidel, Kluwer Academic Publishers, Dordrecht, ISBN 90-277-1296-4.
- Kondepudi, D. (2008). Introduction to Modern Thermodynamics, Wiley, Chichester, ISBN 978-0-470-01598-8.
- Landsberg, P.T. (1978). Thermodynamics and Statistical Mechanics, Oxford University Press, Oxford, ISBN 0-19-851142-6.
- Lewis, G.N., Randall, M. (1923/1961). Thermodynamics, second edition revised by K.S Pitzer, L. Brewer, McGraw-Hill, New York.
- Maxwell, J.C. (1872). Theory of Heat, third edition, Longmans, Green, and Co., London.
- Partington, J.R. (1949). An Advanced Treatise on Physical Chemistry, Volume 1, Fundamental Principles. The Properties of Gases, Longmans, Green, and Co., London.
- Planck, M. (1923/1926). Treatise on Thermodynamics, third English edition translated by A. Ogg from the seventh German edition, Longmans, Green & Co., London.
- Truesdell, C., Bharatha, S. (1977). The Concepts and Logic of Classical Thermodynamics as a Theory of Heat Engines, Rigorously Constructed upon the Foundation Laid by S. Carnot and F. Reech, Springer, New York, ISBN 0-387-07971-8.
| Instrumentation | |
|---|---|
| Techniques | |
| Sampling | |
| Calibration | |
| Prominent publications | |
| |
External links
| Wikisource has the text of the 1911 Encyclopædia Britannica article Calorimetry. |