Clostridium difficile toxin B
Clostridium difficile toxin B is a cytotoxin produced by the bacteria Clostridium difficile. It is one of two major kinds of toxins produced by C. difficile , the other being an enterotoxin (Toxin A). Both are very potent and lethal.[2][3]
| Toxin B | |||||||
|---|---|---|---|---|---|---|---|
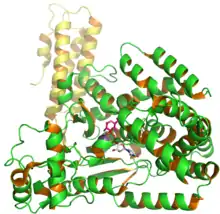 Structure of C. difficile glucosyl transferase Toxin B showing UDP and glusose from PDB entry 2BVM.[1] | |||||||
| Identifiers | |||||||
| Organism | |||||||
| Symbol | toxB | ||||||
| Alt. symbols | tcdB | ||||||
| Entrez | 4914074 | ||||||
| PDB | 2BVM | ||||||
| RefSeq (Prot) | YP_001087135.1 | ||||||
| UniProt | P18177 | ||||||
| Other data | |||||||
| EC number | 2.4.1.- | ||||||
| Chromosome | genome: 0.79 - 0.8 Mb | ||||||
| |||||||
| TcdB toxin N-terminal helical domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | TcdB_N | ||||||||
| Pfam | PF12918 | ||||||||
| |||||||||
| TcdA/TcdB catalytic glycosyltransferase domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | TcdA_TcdB | ||||||||
| Pfam | PF12919 | ||||||||
| |||||||||
| Peptidase C80 family | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | Peptidase_C80 | ||||||||
| Pfam | PF11713 | ||||||||
| InterPro | IPR020974 | ||||||||
| |||||||||
| TcdA/TcdB pore forming domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | TcdA_TcdB_pore | ||||||||
| Pfam | PF12920 | ||||||||
| TCDB | 1.C.57 | ||||||||
| OPM superfamily | 199 | ||||||||
| OPM protein | 6oq6 | ||||||||
| |||||||||
Structure
Toxin B (TcdB) is a cytotoxin that has a molecular weight of 270 kDa and an isoelectric point, pl, of 4.1.[4] Toxin B has four different structural domains: catalytic, cysteine protease, translocation, and receptor binding.[5] The N-terminal glucosyltransferase catalytic domain includes amino acid residues 1–544 while the cysteine protease domain includes residues 545–801. Additionally, the translocation region incorporates amino acid residues from 802 to 1664 while the receptor binding region is part of the C-terminal region and includes amino acid residues from 1665 to 2366.[5]
The glycosylation activity of toxin B occurs in the N-terminal catalytic region (residues 1–544). This region glycosylates substrates independent of any cytotoxic activity.[6] However, a small deletion of the receptor binding region causes attenuation of toxin B activity.[6] The translocation region contains a hydrophobic stalk-like structure, which may help residues 958–1130 in forming membrane spanning pores.[5] The receptor binding region that includes the C-terminal repetitive region (CRR) increases the TcdB membrane interaction but does not participate in pore formation.[7] In addition, cysteine protease and translocation regions both have complex structures that play an important functional role in translocation and receptor binding.[8] However, deleting the translocation region of amino acids decreases the cytotoxic activity 4-fold. Both cysteine proteases and a majority of translocation regions harbor hydrophobic proteins, which show access to TcdB and other toxins crossing the cell membranes.[8]
Receptor binding domain
The C-terminal of TcdB (the green region of Fig. 2) contains a region known as the combined repetitive oligopeptides (CROPs) that contains amino acid residues 1831–2366.[9] These CROPs make up 19–24 short repeats (SRs) of amino acids, roughly 31 long repeats (LRs) of amino acids, toxin A, and Toxin B.[9][10] The TcdB CROPs region consists of 19 SRs and 4 LRs. This SRs and LRs region allows formation of cell wall binding motifs that help to bind sugar moieties of the cell surfaces.[9]
Purification
In order to purify toxin B from C. difficile cell cultures, brain heart infusion broth is used because it promotes the synthesis of toxin B.[11] The filtration method facilitates purification of toxin B from the supernatant of C. difficile. The toxin concentration of the supernatant is proportional to the organism cell count. It has been proposed by many studies that the majority of the toxins are released in either late log phase or early stationary phases, hence, toxin B is continuously secreted by cells.[2] Although there are many methods employed by different studies in purifying toxin B, the majority of studies use methods involving concentrations of ultrafiltrated ammonium sulfate or precipitation, in lieu by either gel filtration or ion-exchange chromatography. In addition, the effectiveness of the ion-exchange chromatography method helps to differentiate between TcdA and TcdB.
Function
When the catalytic threonine residue of glucosyltransferase deactivates a family of small GTPases,e.g. the Rho family; Rac, and Cdc42 inside the target cells disturb signal transduction mechanisms, which leads to dysfunctioning of actin cytoskeleton, cell-cell junction, and apoptosis (Fig. 5).[12][13][14] Rho induces the activity of actin stress fibers. Rac proteins controls the activities of membrane ruffling and NADPH-oxidase neutrophil. Cdc42 regulates the F-actin filament formation in filopodia.
Cytotoxicity
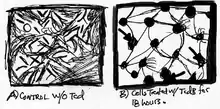
Several studies have demonstrated that the presence of TcdB in mammalian cells leads to rapid changes within cell morphology and cell signaling. Within a short period of time, cells have the appearance of plaque with small dosages of TcdB and TcdA. In addition, death of the cells is a major impact of these toxins after cells have been intoxicated. An investigation by Donta et al., forwarded that TcdB has serious impacts in other mammalian cells such as chinese hamster ovary cells, human cervical epithelial cells, mouse adrenal cells, rat hepatocytes and rat astrocytes (Fig.3).[15][16]
The cytotoxic activity is based on cell types, which could range from 4-fold to 200-fold. Generally, when cells are infected with TcdB, they not only lose their structural integrity, but also diminutions of F-actin filaments.[17] Cell roundings by TcdB take no longer than 2 hours (Fig. 4), but as far as cell death goes, it can take approximately 24 hours.[15] With regard to Clostridium difficile-associated diarrhea (CDAD), the effects of cytopathicity are more critical than actual cell death because once cells lose integrity of the cytoskeleton actin filament, they also lose its normal function.

Effects on small GTPases
The cause of cytotoxic activity by TcdB within the host cell is mainly mediated via receptor endocytosis. Acidic endosomes allow toxin B to enter the cytosol. This phenomenon takes place by a binding receptor region, which enables toxin to enter host cells. Through the accessibility of the host cells' cytosol, TcdB deactivates the small GTPases (Fig. 5), e.g. the Rho family members Rac and Cdc42 by the process of glycosylation of threonine 35 in Cdc42 and Rac, and threonine 37 in Rho.[18][19] These Rho GTPases are found ubiquitously in the cytosol of eukaryotic cells that are responsible for the organization of the actin cytoskeleton because the toxins in the cytosol cause condensation of actin filaments as a consequence of cell rounding and membrane blebbing (Fig. 3), which ultimately leads to apoptosis.[20][21] TcdB causes critical changes to cell dynamics and morphology. Figure 3 shows the probable effect of toxin B on a cell's surface; membrane blebbing (black arrows).[22] In addition, TcdB inactivates Rho GTPases. As a consequence, cell-cell junctions are disrupted, which enhances epithelial permeability of toxin B and fluid accumulation in the lumen. This is one of the main causative agents in contracting Clostridium difficile-associated diarrhea (CDAD)(Fig. 5).[23][24]
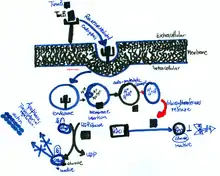
Furthermore, the rate of hydrolysis by TcdB of UDP-glucose is approximately five-fold greater than TcdA.[25] Several studies have indicated that Rho exhibits posttranslational modification through prenylation and carboxymethylation, which occurs in the cytoplasmic side of the plasma membrane, hence, the exchange of GTP to GDP.[26] When TcdB binds to Rho and other small GTPases, GTP hydrolyzes to GDP, which leads to GTP-bound (active) to GDP-bound (inactive) (Fig. 5). In addition, this interchange activity is regulated by guanine factors in the cell's cytosol.[27]
Disturbance on signal pathways
The cellular regulation of Rho, Rac, and Cdc42 has effects outside the vicinity of the actin filaments of the cytoskeleton (Fig. 4),[17] These small GTPases are incorporated in the cell cycle that regulates signals via mitogen-activated protein kinase kinases (MAPKKs).[28] Some physiological parts of the cells that are not involved in actin filaments, may not cause cell rounding or cell death right away, but in downstream pathway activity, may lead to the deterioration of actin filaments and finally, cell death.[17]
In 1993, a study conducted by Shoshan et al., showed that cells with TcdB changed phospholipase A2 activity. This was an independent event from disruption of the actin cytoskeleton.[29] Shoshan et al., also showed that TcdB inhibited the receptor signaling activity by deactivating the Rho proteins via phospholipase D.[29]
Pore formation
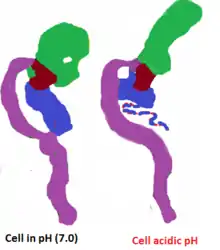
TcdB accesses the interior of the cell through clathrin-mediated endocytosis,[30] When toxin B is part of the cytosol, the glucosyltransferase passes through the endosomal membrane, which decreases pH, induces translocation and finally leads to morphological changes of translocation region residues (958–1130).[31] The hydrophobic regions are embedded in the host membrane to form pores that allow glucosyltransferase domains to pass through.[31] When cells are infected with TcdB in an acidic environment, it attenuates toxins and causes shape rearrangements (Fig. 6).[31] As a consequence of acidic pH, TcdB displays clear differences in original fluorescence of tryptophan, susceptibility of proteases, and hydrophobic surfaces.[31] Another group has shown that acidification leads to conformational changes of the toxin and, more importantly, helps to form pores.[7] A putative translocation region ( Fig. 2) makes up approximately 801–1400 amino acids, of which residues 958–1130 are hydrophobic and are responsible for the formation of transmembrane pores.[20] A majority of the studies used TcdB strain 630 to show the pore formation activity of C. difficile toxins.[31]
Induced by pH
To see whether effects of proteolytic cleavage of TcdB takes place at the cell surface or in acidic endosomes, studies used Bafilomycin A1, which is known to block the v-type H+-ATPases of endosomes. This reduces the acidity in endosomes.[31] The physiological uptake pathway of TcdB prevents cytopathic activity by TcdB.[31] When cells were in acidic conditions (pH 4.0) for 5 minutes after binding TcdB to the cell surface at 37 degree Celsius, the shape rearrangements and rounding were observed. However, when rounded cells were incubated for an additional hour in neutral pH (7.0) with similar parameters, no cell rounding was observed.[15][31] Both studies showed that toxin B has a property of proteolytic cleavage, which is critical for access to the cytosol.[7][15][31] Having an acidic endosome pH leads to topological alterations of TcdB (Figure 6).[7]
Genetics
The gene that encodes the TcdB protein, tcdB, is located within the chromosomal region of 19.6 kb. This is known as the locus of pathogenicity or PaLoc (Figure 2).[32][33] The open reading frame (ORF) for tcdB is 7,098 nucleotides in length.[17] It is important to mention that—besides the major toxin genes in the PaLoc region—there are three other accessory genes that encode in the PaLoc region: tcdR (L), tcdC (R) and tcdE in the middle. These genes help to regulate TcdA and TcdB expression. They also help to secrete or release the toxins from the cell.[17] The encoding gene tcdE, located between tcdB and tcdA, is analogous to holin proteins, thus, it is suggested that tcdE works as a facilitator gene that enhances the release or secretion of TcdA and TcdB consequently increasing the permeability of the host cell membrane.[17]
%252Cencoding_the_large_clostridial_toxins(LCTs)_involved_in_C._difficile_infections_CDI.png.webp)
Toxin detection
There are different plasmid sizes of C. difficile. The detected molecular weights range from 2.7x106 to 100x106, but plasmid sizes show no correlation with toxicity. In order to detect the toxin B level in C. difficile, clinicians extensively use cell culture assays derived from stool specimens from patients with PMC.[2][3] The cell culture assay is regarded as a “gold standard” for detecting toxicity in C. difficile because a small quantity of toxin B is capable of causing cell rounding (Fig. 4), thus, it is a major advantage of clinical laboratories to make correlations with the CDAD caused by TcdB.[2][3] Although cytotoxic activity of large clostridial toxins (LCTs) was found in PMC patient stool specimens, toxin B activity had more detrimental cytotoxic effects in comparison with toxin A.[2] Therefore, the activity of toxin A is attenuated when it is not isolated from toxin B.[2][3] The detection of C. difficile toxicity is extremely sensitive, however, using the cell culture assay allows clinical laboratories to overcome the challenge; using doses as little as 1 pg/mL of toxin B is enough to causes cell rounding.[2][3] This is the major advantage in using the culture tissue assay to detect toxicity in PMC patients.[2] Even though clinical laboratories have tried to use an assay microtiter plate enzyme-linked immunosorbent assay (ELISA) and other techniques to detect the cytotoxic activity of toxin B in the feces of PMC patients, the results are not as accurate as those where cell culture assays were used.[2][3][34]
Production factor
By adding antimicrobial, e.g. clindamycin, into the culture growth medium, studies have shown that the cytotoxic activity in C. difficile cultures increases by 4–8 fold.[35][36] Moreover, knowing the role of antibiotics on the causes of PMC, many earlier studies focused on the effects of antimicrobials production of toxins. As a result, studies were able to conclude that the subinhibitory nature of vancomycin and penicillin levels were increasing the toxin production in cultures of C. difficile.[37] The amounts of toxin production were correlated with the usage of growth medium for the organisms. Another study illustrated that the high levels of toxin production of TcdB were observed in complex mediums such as brain and heart infusion broth.[38][39] High levels of toxins were produced with isolation of highly virulent. Conversely, low levels of toxins were produced with isolation of weakly virulent. Thus, it shows that the productions of toxins were co-regulated. Although the mechanism behind the environment's involvement in modulating the signals expressing the toxins is not understood, in vitro studies have shown that expression of toxin is strengthened by catabolite repression and stress, e.g. antibiotics.[40][41][42] Another study has shown that limiting biotin in well-characterized medium increases the production of TcdB by 64-fold and TcdA by 35-fold. This was done with C. difficile and doses of biotin as small as 0.05nM.[41] Several other early studies have argued against the theory that the production of toxin has anything to deal with stress or catabolite repression of either toxin TcdA or TcdB.[42] Also, many studies say the main reason for the differences among other studies is due to toxin production not occurring with all isolates of C. difficile.
Clinical significance
Many early studies have suggested that toxin A (also known as TcdA) is the major toxin protein causing antibiotic-associated diarrhea (AAD); however, research scientists within the last decade or so have shown that Toxin B (or TcdB) plays a more important role in disease than anyone had forecasted. With this knowledge, Toxin B has been identified as the major virulence factor that causes the opening of tight junctions of intestinal epithelial cells, which enables toxin to increase vascular permeability and induce hemorrhaging. Hence, this leads to tumor necrosis factor α (TNF α) and proinflammatory interleukins being established as the major causative agents of pseudomembranous colitis (PMC) and antibiotic-associated diarrhea (AAD).[2][3][43]
The involvement of toxin A and—most importantly—toxin B is the key element that determines the disease caused by C. difficile. Clinical laboratories have identified these toxins in patients' stool based on antibody and cytotoxicity assays.[44] These bacterial toxins have been shown to be associated with Clostridium sordellii hemorrhagic toxin (TcsH), lethal toxin (TcsL), and Clostridium novyi alpha toxin (Tcn α), thus, making this cohort to be the large family of toxin clostridial.[17] Because of similarities of these toxins with others, researchers have classified them as the family of large clostridial toxins (LCTs).[9]
Mechanism of bezlotoxumab with TcdB
Bezlotoxumab is a human monoclonal antibody designed for the prevention of recurrence of Clostridium difficile infections. By x-ray crystallized structure of N-terminal of TcdB, the toxin is identified to consist of three domains: a glucosyltransferase domain (GTD), a cysteine protease and a combined repetitive oligopeptide (CROP) domain. Bezlotoxumab specifically binds to two homologous sites within the CROP domain of TcdB. Structural analysis by X-ray crystallography indicates that antibody binding partially occludes putative carbohydrate binding pockets. Consistent with this idea, Bezlotoxumab blocks binding of TcdB to mammalian cells.[45]
Role in pseudomembranous colitis
In early stages of the disease of PMC, many studies have speculated that TcdA is more potent than TcdB. This has been deduced from in vivo experiments where toxin productions of TcdA were more severe than TcdB with antibiotics cecitis.[38][46] Later, several studies showed that TcdB plays a major role in the disease of PMC and ADD. The study demonstrated that even though C. difficile did not produces TcdA, it still showed symptoms for the disease.[47] Furthermore, later studies have shown that a purified form of TcdB is a more lethal enterotoxin in comparison to TcdA, and also, that intestinal epithelium is severely damaged and causes an acute inflammatory response.[48] With better understanding of the toxin, researchers were able to state that TcdB is the major virulence factor that causes CDI over TcdA. However, when TcdA is present in the gut, it helps to facilitate TcdB's activity to have broader impacts, consequently, affecting multiple organ systems.[49] In addition, when hamsters were vaccinated against TcdA, it showed hamsters were not fully protected from the C. difficile disease and this lead studies to conclude that TcdB is very lethal and potent.[50] Furthermore, injecting a small dose of TcdA with a lethal dose of TcdB intravenously or intraperitoneally proved sufficient in causing the death of an animal. Therefore, TcdA works as a facilitator of TcdB exiting from the gut.[50]
See also
References
- Reinert DJ, Jank T, Aktories K, Schulz GE (September 2005). "Structural basis for the function of Clostridium difficile toxin B". Journal of Molecular Biology. 351 (5): 973–81. doi:10.1016/j.jmb.2005.06.071. PMID 16054646.
- Lyerly DM, Krivan HC, Wilkins TD (January 1988). "Clostridium difficile: its disease and toxins". Clinical Microbiology Reviews. 1 (1): 1–18. doi:10.1128/cmr.1.1.1. PMC 358025. PMID 3144429.
- Bartlett JG (1990). "Clostridium difficile: clinical considerations". Reviews of Infectious Diseases. 12 Suppl 2: S243–51. doi:10.1093/clinids/12.Supplement_2.S243. PMID 2406876.
- von Eichel-Streiber C (1997). "Enterotoxin A and cytotoxin B (Clostridium difficile)". In Montecucco C, Rappuoli R (eds.). Guidebook to protein toxins and their use in cell biology. Oxford [Oxfordshire]: Oxford University Press. p. 72. ISBN 978-0-19-859954-8.
- Albesa-Jové D, Bertrand T, Carpenter EP, Swain GV, Lim J, Zhang J, Haire LF, Vasisht N, Braun V, Lange A, von Eichel-Streiber C, Svergun DI, Fairweather NF, Brown KA (March 2010). "Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS". Journal of Molecular Biology. 396 (5): 1260–70. doi:10.1016/j.jmb.2010.01.012. PMID 20070948.
- Hofmann F, Busch C, Prepens U, Just I, Aktories K (April 1997). "Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin". Journal of Biological Chemistry. 272 (17): 11074–8. doi:10.1074/jbc.272.17.11074. PMID 9111001.
- Barth H, Pfeifer G, Hofmann F, Maier E, Benz R, Aktories K (April 2001). "Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells". Journal of Biological Chemistry. 276 (14): 10670–6. doi:10.1074/jbc.M009445200. PMID 11152463.
- Jank T, Aktories K (May 2008). "Structure and mode of action of clostridial glucosylating toxins: the ABCD model". Trends in Microbiology. 16 (5): 222–9. doi:10.1016/j.tim.2008.01.011. PMID 18394902.
- von Eichel-Streiber C, Boquet P, Sauerborn M, Thelestam M (October 1996). "Large clostridial cytotoxins--a family of glycosyltransferases modifying small GTP-binding proteins". Trends in Microbiology. 4 (10): 375–82. doi:10.1016/0966-842X(96)10061-5. PMID 8899962.
- Jank T, Giesemann T, Aktories K (April 2007). "Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function". Glycobiology. 17 (4): 15R–22R. doi:10.1093/glycob/cwm004. PMID 17237138.
- Meador J, Tweten RK (July 1988). "Purification and characterization of toxin B from Clostridium difficile". Infection and Immunity. 56 (7): 1708–14. PMC 259466. PMID 3384474.
- Aktories K, Just I (December 1995). "Monoglucosylation of low-molecular-mass GTP-binding Rho proteins by clostridial cytotoxins". Trends in Cell Biology. 5 (12): 441–3. doi:10.1016/S0962-8924(00)89107-2. PMID 14732022.
- Dillon ST, Rubin EJ, Yakubovich M, Pothoulakis C, LaMont JT, Feig LA, Gilbert RJ (April 1995). "Involvement of Ras-related Rho proteins in the mechanisms of action of Clostridium difficile toxin A and toxin B". Infection and Immunity. 63 (4): 1421–6. PMC 173169. PMID 7890404.
- Wilkins TD, Lyerly DM (February 1996). "Clostridium difficile toxins attack Rho". Trends in Microbiology. 4 (2): 49–51. doi:10.1016/0966-842X(96)81508-3. PMID 8820565.
- Pfeifer G, Schirmer J, Leemhuis J, Busch C, Meyer DK, Aktories K, Barth H (November 2003). "Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells". Journal of Biological Chemistry. 278 (45): 44535–41. doi:10.1074/jbc.M307540200. PMID 12941936.
- Donta ST, Sullivan N, Wilkins TD (June 1982). "Differential effects of Clostridium difficile toxins on tissue-cultured cells". Journal of Clinical Microbiology. 15 (6): 1157–8. PMC 272271. PMID 7107845.
- Voth DE, Ballard JD (April 2005). "Clostridium difficile toxins: mechanism of action and role in disease". Clinical Microbiology Reviews. 18 (2): 247–63. doi:10.1128/CMR.18.2.247-263.2005. PMC 1082799. PMID 15831824.
- Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K (June 1995). "Glucosylation of Rho proteins by Clostridium difficile toxin B". Nature. 375 (6531): 500–3. doi:10.1038/375500a0. PMID 7777059.
- Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K (June 1995). "The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins". Journal of Biological Chemistry. 270 (23): 13932–6. doi:10.1074/jbc.270.23.13932. PMID 7775453.
- von Eichel-Streiber C, Warfolomeow I, Knautz D, Sauerborn M, Hadding U (November 1991). "Morphological changes in adherent cells induced by Clostridium difficile toxins" (PDF). Biochemical Society Transactions. 19 (4): 1154–60. doi:10.1042/bst0191154. PMID 1794484.
- Thelestam M, Chaves-Olarte E (2000). Cytotoxic effects of the Clostridium difficile toxins. Current Topics in Microbiology and Immunology. 250. pp. 85–96. doi:10.1007/978-3-662-06272-2_4. ISBN 978-3-642-08668-7. PMID 10981358.
- Fiorentini C, Fabbri A, Falzano L, Fattorossi A, Matarrese P, Rivabene R, Donelli G (June 1998). "Clostridium difficile toxin B induces apoptosis in intestinal cultured cells". Infection and Immunity. 66 (6): 2660–5. PMC 108253. PMID 9596731.
- Feltis BA, Wiesner SM, Kim AS, Erlandsen SL, Lyerly DL, Wilkins TD, Wells CL (December 2000). "Clostridium difficile toxins A and B can alter epithelial permeability and promote bacterial paracellular migration through HT-29 enterocytes". Shock. 14 (6): 629–34. doi:10.1097/00024382-200014060-00010. PMID 11131913.
- Johal SS, Solomon K, Dodson S, Borriello SP, Mahida YR (June 2004). "Differential effects of varying concentrations of Clostridium difficile toxin A on epithelial barrier function and expression of cytokines". The Journal of Infectious Diseases. 189 (11): 2110–9. doi:10.1086/386287. PMID 15143480.
- Ciesla WP, Bobak DA (June 1998). "Clostridium difficile toxins A and B are cation-dependent UDP-glucose hydrolases with differing catalytic activities". Journal of Biological Chemistry. 273 (26): 16021–6. doi:10.1074/jbc.273.26.16021. PMID 9632652.
- Adamson P, Marshall CJ, Hall A, Tilbrook PA (October 1992). "Post-translational modifications of p21rho proteins". Journal of Biological Chemistry. 267 (28): 20033–8. PMID 1400319.
- Zhou K, Wang Y, Gorski JL, Nomura N, Collard J, Bokoch GM (July 1998). "Guanine nucleotide exchange factors regulate specificity of downstream signaling from Rac and Cdc42". Journal of Biological Chemistry. 273 (27): 16782–6. doi:10.1074/jbc.273.27.16782. PMID 9642235.
- Zhang Y, Dong C (November 2007). "Regulatory mechanisms of mitogen-activated kinase signaling". Cellular and Molecular Life Sciences. 64 (21): 2771–89. doi:10.1007/s00018-007-7012-3. PMID 17726577.
- Shoshan MC, Florin I, Thelestam M (May 1993). "Activation of cellular phospholipase A2 by Clostridium difficile toxin B". Journal of Cellular Biochemistry. 52 (1): 116–24. doi:10.1002/jcb.240520115. PMID 8320270.
- Papatheodorou P, Zamboglou C, Genisyuerek S, Guttenberg G, Aktories K (May 2010). "Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis". PLOS ONE. 5 (5): e10673. doi:10.1371/journal.pone.0010673. PMC 2871790. PMID 20498856.
- Qa'Dan M, Spyres LM, Ballard JD (May 2000). "pH-induced conformational changes in Clostridium difficile toxin B". Infection and Immunity. 68 (5): 2470–4. doi:10.1128/IAI.68.5.2470-2474.2000. PMC 97448. PMID 10768933.
- Carter GP, Rood JI, Lyras D (January 2012). "The role of toxin A and toxin B in the virulence of Clostridium difficile". Trends in Microbiology. 20 (1): 21–9. doi:10.1016/j.tim.2011.11.003. PMID 22154163.
- Braun V, Hundsberger T, Leukel P, Sauerborn M, von Eichel-Streiber C (November 1996). "Definition of the single integration site of the pathogenicity locus in Clostridium difficile". Gene. 181 (1–2): 29–38. doi:10.1016/S0378-1119(96)00398-8. PMID 8973304.
- Musher DM, Manhas A, Jain P, Nuila F, Waqar A, Logan N, Marino B, Graviss EA (August 2007). "Detection of Clostridium difficile toxin: comparison of enzyme immunoassay results with results obtained by cytotoxicity assay". Journal of Clinical Microbiology. 45 (8): 2737–9. doi:10.1128/JCM.00686-07. PMC 1951241. PMID 17567791.
- Nakamura S, Mikawa M, Tanabe N, Yamakawa K, Nishida S (1982). "Effect of clindamycin on cytotoxin production by Clostridium difficile". Microbiology and Immunology. 26 (11): 985–92. doi:10.1111/j.1348-0421.1982.tb00248.x. PMID 7167065.
- George RH, Johnson M, Youngs D, Burdon DW (1980). "Induction of Clostridium difficile toxin by antibiotics". Current Chemotherapy and Infectious Diseases. 2 (1): 955–56.
- Onderdonk AB, Lowe BR, Bartlett JG (October 1979). "Effect of environmental stress on Clostridium difficile toxin levels during continuous cultivation". Applied and Environmental Microbiology. 38 (4): 637–41. PMC 243552. PMID 44176.
- Lyerly DM, Sullivan NM, Wilkins TD (January 1983). "Enzyme-linked immunosorbent assay for Clostridium difficile toxin A". Journal of Clinical Microbiology. 17 (1): 72–8. PMC 272577. PMID 6338036.
- Sullivan NM, Pellett S, Wilkins TD (March 1982). "Purification and characterization of toxins A and B of Clostridium difficile". Infection and Immunity. 35 (3): 1032–40. PMC 351151. PMID 7068210.
- Dupuy B, Sonenshein AL (January 1998). "Regulated transcription of Clostridium difficile toxin genes". Molecular Microbiology. 27 (1): 107–20. doi:10.1046/j.1365-2958.1998.00663.x. PMID 9466260.
- Yamakawa K, Karasawa T, Ikoma S, Nakamura S (February 1996). "Enhancement of Clostridium difficile toxin production in biotin-limited conditions". Journal of Medical Microbiology. 44 (2): 111–4. CiteSeerX 10.1.1.623.71. doi:10.1099/00222615-44-2-111. PMID 8642571.
- Mani N, Dupuy B (May 2001). "Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor". Proceedings of the National Academy of Sciences of the United States of America. 98 (10): 5844–9. doi:10.1073/pnas.101126598. PMC 33301. PMID 11320220.
- Bartlett JG (May 1994). "Clostridium difficile: history of its role as an enteric pathogen and the current state of knowledge about the organism". Clinical Infectious Diseases. 18 Suppl 4: S265–72. doi:10.1093/clinids/18.Supplement_4.S265. PMID 8086574.
- Carter GP, Rood JI, Lyras D (January 2012). "The role of toxin A and toxin B in the virulence of Clostridium difficile". Trends in Microbiology. 20 (1): 21–9. doi:10.1016/j.tim.2011.11.003. PMID 22154163.
- Orth P, Xiao L, Hernandez LD, Reichert P, Sheth PR, Beaumont M, et al. (June 2014). "Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography". The Journal of Biological Chemistry. 289 (26): 18008–21. doi:10.1074/jbc.m114.560748. PMC 4140266. PMID 24821719.
- Arnon SS, Mills DC, Day PA, Henrickson RV, Sullivan NM, Wilkins TD (January 1984). "Rapid death of infant rhesus monkeys injected with Clostridium difficile toxins A and B: physiologic and pathologic basis". Journal of Pediatrics. 104 (1): 34–40. doi:10.1016/S0022-3476(84)80585-5. PMID 6690674.
- Drudy D, Fanning S, Kyne L (January 2007). "Toxin A-negative, toxin B-positive Clostridium difficile". The Journal of Infectious Diseases. 11 (1): 5–10. doi:10.1016/j.ijid.2006.04.003. PMID 16857405.
- Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C (August 2003). "Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine". Gastroenterology. 125 (2): 413–20. doi:10.1016/S0016-5085(03)00902-8. PMID 12891543.
- Dobson G, Hickey C, Trinder J (June 2003). "Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome". Intensive Care Medicine. 29 (6): 1030. doi:10.1007/s00134-003-1754-7. PMID 12734650.
- Lyerly, DM; Roberts, MD; Phelps, CJ; Wilkins, TD (January 1986). "Purification and properties of toxins A and B of Clostridium difficile". FEMS Microbiology Letters. 33 (1): 31–35. doi:10.1111/j.1574-6968.1986.tb01206.x.