Creutzfeldt–Jakob disease
Creutzfeldt–Jakob disease (CJD), also known as subacute spongiform encephalopathy or neurocognitive disorder due to prion disease, is a fatal degenerative brain disorder.[4][1] Early symptoms include memory problems, behavioral changes, poor coordination, and visual disturbances.[4] Later symptoms include dementia, involuntary movements, blindness, weakness, and coma.[4] About 70% of people die within a year of diagnosis.[4]
| Creutzfeldt–Jakob disease | |
|---|---|
| Other names | Classic Creutzfeldt–Jakob disease[1] |
 | |
| Magnetic resonance image of sporadic CJD[2] | |
| Pronunciation | |
| Specialty | Neurology |
| Symptoms | Early: memory problems, behavioral changes, poor coordination, visual disturbances[4] Later: dementia, involuntary movements, blindness, weakness, coma[4] |
| Complications | Aspiration pneumonia due to difficulty coughing |
| Usual onset | Around 60[4] |
| Duration | 70% die within a year of diagnosis[4] |
| Types | Sporadic, hereditary, acquired, Variant[4] |
| Causes | Prion[4] |
| Risk factors | Having at least one living or deceased ancestor with the disease (in case of hereditary CJD) |
| Diagnostic method | Based on symptoms and medical tests after other possible causes are ruled out[4] |
| Differential diagnosis | Encephalitis, chronic meningitis, Huntington’s disease, Alzheimer's disease[4] |
| Treatment | Supportive care[4] |
| Medication | For pain relief: Morphine, Methadone |
| Prognosis | Universally fatal[4] |
| Frequency | 1 per million per year[4] |
CJD is caused by a protein known as a prion.[5] Infectious prions are misfolded proteins that can cause normally folded proteins to become misfolded.[4] About 85% of cases occur for unknown reasons, while about 7.5% of cases are inherited from a person's parents in an autosomal dominant manner.[4][6] Exposure to brain or spinal tissue from an infected person may also result in spread.[4] There is no evidence that it can spread between people via normal contact or blood transfusions.[4] Diagnosis involves ruling out other potential causes.[4] An electroencephalogram, spinal tap, or magnetic resonance imaging may support the diagnosis.[4]
There is no specific treatment for CJD.[4] Opioids may be used to help with pain, while clonazepam or sodium valproate may help with involuntary movements.[4] CJD affects about one per million people per year.[4] Onset is typically around 60 years of age.[4] The condition was first described in 1920.[4] It is classified as a type of transmissible spongiform encephalopathy.[7] Inherited CJD accounts for about 10% of prion disease cases.[6] Sporadic CJD is different from bovine spongiform encephalopathy (mad cow disease) and variant Creutzfeldt–Jakob disease (vCJD).[8]
Signs and symptoms
The first symptom of CJD is usually rapidly progressive dementia, leading to memory loss, personality changes, and hallucinations. Myoclonus (jerky movements) typically occurs in 90% of cases, but may be absent at initial onset.[9] Other frequently occurring features include anxiety, depression, paranoia, obsessive-compulsive symptoms, and psychosis.[10] This is accompanied by physical problems such as speech impairment, balance and coordination dysfunction (ataxia), changes in gait, and rigid posture. In most people with CJD, these symptoms are accompanied by involuntary movements. The duration of the disease varies greatly, but sporadic (non-inherited) CJD can be fatal within months or even weeks.[11] Most victims die six months after initial symptoms appear, often of pneumonia due to impaired coughing reflexes. About 15% of people with CJD survive for two or more years.[12]
The symptoms of CJD are caused by the progressive death of the brain's nerve cells, which are associated with the build-up of abnormal prion proteins forming in the brain. When brain tissue from a person with CJD is examined under a microscope, many tiny holes can be seen where the nerve cells have died. Parts of the brain may resemble a sponge where the prion were infecting the areas of the brain.[13]
Cause
CJD is a type of transmissible spongiform encephalopathy (TSE), which are caused by prions.[5] Prions are misfolded proteins that occur in the neurons of the central nervous system (CNS). They are thought to affect signaling processes, damaging neurons and resulting in degeneration that causes the spongiform appearance in the affected brain.[14]
The CJD prion is dangerous because it promotes refolding of native prion protein into the diseased state.[15] The number of misfolded protein molecules will increase exponentially and the process leads to a large quantity of insoluble protein in affected cells. This mass of misfolded proteins disrupts neuronal cell function and causes cell death. Mutations in the gene for the prion protein can cause a misfolding of the dominantly alpha helical regions into beta pleated sheets. This change in conformation disables the ability of the protein to undergo digestion. Once the prion is transmitted, the defective proteins invade the brain and induce other prion protein molecules to misfold in a self-sustaining feedback loop. These neurodegenerative diseases are commonly called prion diseases.
People can also develop CJD because they carry a mutation of the gene that codes for the prion protein (PRNP). This occurs in only 5–10% of all CJD cases. In sporadic cases, the misfolding of the prion protein is a process which is hypothesized to occur as a result of the effects of aging on cellular machinery, explaining why the disease often appears later in life.[4][16] An EU study determined that "87% of cases were sporadic, 8% genetic, 5% iatrogenic and less than 1% variant."[17]
Transmission
The defective protein can be transmitted by contaminated harvested human brain products, corneal grafts,[18] dural grafts,[19] or electrode[20] implants and human growth hormone.[21]
It can be familial (fCJD); or it may appear without clear risk factors (sporadic form: sCJD). In the familial form, a mutation has occurred in the gene for PrP, PRNP, in that family. All types of CJD are transmissible irrespective of how they occur in the person.[22]
It is thought that humans can contract the variant form of the disease by eating food from animals infected with bovine spongiform encephalopathy (BSE), the bovine form of TSE also known as mad cow disease. However, it can also cause sCJD in some cases.[23][24]
Cannibalism has also been implicated as a transmission mechanism for abnormal prions, causing the disease known as kuru, once found primarily among women and children of the Fore people in Papua New Guinea.[25] While the men of the tribe ate the muscle tissue of the deceased, women and children consumed other parts, such as the brain, and were more likely than men to contract kuru from infected tissue.
Prions, the infectious agent of CJD, may not be inactivated by means of routine surgical instrument sterilization procedures. The World Health Organization and the US Centers for Disease Control and Prevention recommend that instrumentation used in such cases be immediately destroyed after use; short of destruction, it is recommended that heat and chemical decontamination be used in combination to process instruments that come in contact with high-infectivity tissues. No cases of health care associated transmission of CJD have been reported subsequent to the adoption of current sterilization procedures, or since 1976.[26][27][28] Copper-hydrogen peroxide has been suggested as an alternative to the current recommendation of sodium hydroxide or sodium hypochlorite.[29] Thermal depolymerization also destroys prions in infected organic and inorganic matter, since the process chemically attacks protein at the molecular level, although more effective and practical methods involve destruction by combinations of detergents and enzymes similar to biological washing powders.[30]
Blood products
As of 2018, evidence suggests that while there may be prions in the blood of individuals with vCJD, this is not the case in individuals with sporadic CJD.[4]
Diagnosis
Testing for CJD has historically been problematic, due to nonspecific nature of early symptoms and difficulty in safely obtaining brain tissue for confirmation. The diagnosis may initially be suspected in a person with rapidly progressing dementia, particularly when they are also found with the characteristic medical signs and symptoms such as involuntary muscle jerking,[31] difficulty with coordination/balance and walking, and visual disturbances.[32] Further testing can support the diagnosis and may include:
- Electroencephalography – may have characteristic generalized periodic sharp wave pattern. Periodic sharp wave complexes develop in half of the people with sporadic CJD, particularly in the later stages.[2]
- Cerebrospinal fluid (CSF) analysis for elevated levels of 14-3-3 protein could be supportive in the diagnosis of sCJD. However, a positive result should not be regarded as sufficient for the diagnosis.[33][34][35] The Real-Time Quaking-Induced Conversion (RT-QuIC) assay has a diagnostic sensitivity of more than 80% and a specificity approaching 100%, tested in detecting PrPSc in CSF samples of people with CJD.[36][37][38] It is therefore suggested as a high-value diagnostic method for the disease.
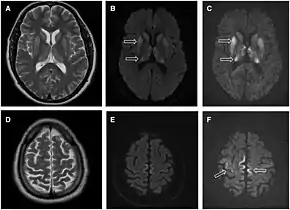
- MRI of the brain – often shows high signal intensity in the caudate nucleus and putamen bilaterally on T2-weighted images.
In recent years, studies have shown that the tumour marker Neuron-specific enolase (NSE) is often elevated in CJD cases; however, its diagnostic utility is seen primarily when combined with a test for the 14-3-3 protein.[39] As of 2010, screening tests to identify infected asymptomatic individuals, such as blood donors, are not yet available, though methods have been proposed and evaluated.[40]
Imaging
Imaging of the brain may be performed during medical evaluation, both to rule out other causes and to obtain supportive evidence for diagnosis. Imaging findings are variable in their appearance, and also variable in sensitivity and specificity.[41] While imaging plays a lesser role in diagnosis of CJD,[42] characteristic findings on brain MRI in some cases may precede onset of clinical manifestations.[43]
Brain MRI is the most useful imaging modality for changes related to CJD. Of the MRI sequences, diffuse-weighted imaging sequences are most sensitive. Characteristic findings are as follows:
- Focal or diffuse diffusion-restriction involving the cerebral cortex and/or basal ganglia. In about 24% of cases DWI shows only cortical hyperintensity; in 68%, cortical and subcortical abnormalities; and in 5%, only subcortical anomalies.[44] The most iconic and striking cortical abnormality has been called "cortical ribboning" or "cortical ribbon sign" due to hyperintensities resembling ribbons appearing in the cortex on MRI.[45] The involvement of the thalamus can be found in sCJD, is even stronger and constant in vCJD.[46]
- Varying degree of symmetric T2 hyperintense signal changes in the basal ganglia (i.e., caudate and putamen), and to a lesser extent globus pallidus and occipital cortex.[42]
- Cerebellar atrophy
Histopathology
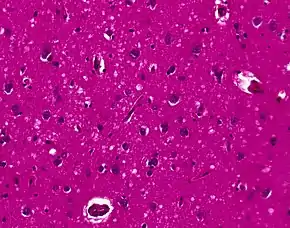
Testing of tissue remains the most definitive way of confirming the diagnosis of CJD, although it must be recognized that even biopsy is not always conclusive.
In one-third of people with sporadic CJD, deposits of "prion protein (scrapie)," PrPSc, can be found in the skeletal muscle and/or the spleen. Diagnosis of vCJD can be supported by biopsy of the tonsils, which harbor significant amounts of PrPSc; however, biopsy of brain tissue is the definitive diagnostic test for all other forms of prion disease. Due to its invasiveness, biopsy will not be done if clinical suspicion is sufficiently high or low. A negative biopsy does not rule out CJD, since it may predominate in a specific part of the brain.[47]
The classic histologic appearance is spongiform change in the gray matter: the presence of many round vacuoles from one to 50 micrometers in the neuropil, in all six cortical layers in the cerebral cortex or with diffuse involvement of the cerebellar molecular layer.[48] These vacuoles appear glassy or eosinophilic and may coalesce. Neuronal loss and gliosis are also seen.[49] Plaques of amyloid-like material can be seen in the neocortex in some cases of CJD.
However, extra-neuronal vacuolization can also be seen in other disease states. Diffuse cortical vacuolization occurs in Alzheimer's disease, and superficial cortical vacuolization occurs in ischemia and frontotemporal dementia. These vacuoles appear clear and punched-out. Larger vacuoles encircling neurons, vessels, and glia are a possible processing artifact.[47]
Classification
Types of CJD include:[50]
- Sporadic (sCJD), caused by the spontaneous misfolding of prion-protein in an individual.[16] This accounts for 85% of cases of CJD.[51]
- Familial (fCJD), caused by an inherited mutation in the prion-protein gene.[50] This accounts for the majority of the other 15% of cases of CJD.[51]
- Acquired CJD, caused by contamination with tissue from an infected person, usually as the result of a medical procedure (iatrogenic CJD). Medical procedures that are associated with the spread of this form of CJD include blood transfusion from the infected person, use of human-derived pituitary growth hormones, gonadotropin hormone therapy, and corneal and meningeal transplants.[50][51][52] Variant Creutzfeldt–Jakob disease (vCJD) is a type of acquired CJD potentially acquired from bovine spongiform encephalopathy or caused by consuming food contaminated with prions.[50][53]
| Characteristic | Classic CJD | Variant CJD |
|---|---|---|
| Median age at death | 68 years | 28 years |
| Median duration of illness | 4–5 months | 13–14 months |
| Clinical signs and symptoms | Dementia; early neurologic signs | Prominent psychiatric/behavioral symptoms; painful dysesthesias; delayed neurologic signs |
| Periodic sharp waves on electroencephalogram | Often present | Often absent |
| Signal hyperintensity in the caudate nucleus and putamen on diffusion-weighted and FLAIR MRI | Often present | Often absent |
| Pulvinar sign-bilateral high signal intensities on axial FLAIR MRI. Also posterior thalamic involvement on sagittal T2 sequences | Not reported | Present in >75% of cases |
| Immunohistochemical analysis of brain tissue | Variable accumulation. | Marked accumulation of protease-resistant prion protein |
| Presence of agent in lymphoid tissue | Not readily detected | Readily detected |
| Increased glycoform ratio on immunoblot analysis of protease-resistant prion protein | Not reported | Marked accumulation of protease-resistant prion protein |
| Presence of amyloid plaques in brain tissue | May be present | May be present |
Treatment
As of 2021, there is no cure or effective treatment for CJD.[55] Some of the symptoms like twitching can be managed, but otherwise treatment is palliative care.[55] Psychiatric symptoms like anxiety and depression can be treated with sedatives and antidepressants. Myoclonic jerks can be handled with clonazepam or sodium valproate. Opiates can help in pain.[56] Seizures are very uncommon but can nevertheless be treated with antiepileptic drugs.[57]
Prognosis
The condition is universally fatal. As of 1981 people are not known to have lived longer than 2.5 years after the onset of CJD symptoms.[58]
Epidemiology
CDC monitors the occurrence of CJD in the United States through periodic reviews of national mortality data. According to the CDC:
- CJD occurs worldwide at a rate of about 1 case per million population per year.
- On the basis of mortality surveillance from 1979 to 1994, the annual incidence of CJD remained stable at approximately 1 case per million people in the United States.
- In the United States, CJD deaths among people younger than 30 years of age are extremely rare (fewer than five deaths per billion per year).[59][60]
- The disease is found most frequently in people 55–65 years of age, but cases can occur in people older than 90 years and younger than 55 years of age.
- In more than 85% of cases, the duration of CJD is less than 1 year (median: four months) after onset of symptoms.[59][60]
Further information from the CDC:
- Risk of developing CJD increases with age.[61]
- CJD incidence was 3.5 cases per million among those over 50 years of age between 1979-2017.[61]
- Approximately 85% of CJD cases are sporadic and 10-15% of CJD cases are due to inherited mutations of the prion protein gene.[61]
- CJD deaths and age-adjusted death rate in the United States indicates an increasing trend in the number of deaths between 1979-2017[61]
Although not fully understood, additional information suggests that CJD rates in African American and nonwhite groups are lower than in whites.[62] While the mean onset is approximately 67 years of age, cases of sCJD have been reported as young as 17 years and over 80 years of age.[62] Mental capabilities rapidly deteriorate and the average amount of time from onset of symptoms to death is 7 to 9 months.[62]
According to a 2020 systematic review on the international epidemiology of CJD:
- Surveillance studies from 2005 and newer show the estimated global incidence is 1-2 cases per million population per year.[63]
- Sporadic CJD (sCJD) incidence increased from the years 1990-2018 in the UK.[63]
- Probable or definite sCJD deaths also increased from the years 1996-2018 in twelve additional countries.[63]
- CJD incidence is greatest in those over the age of 55 years old, with an average age of 67 years old.[63]
The intensity of CJD surveillance increases the number of reported cases, often in countries where CJD epidemics have occurred in the past and where surveillance resources are greatest.[63] An increase in surveillance and reporting of CJD is most likely in response to BSE and vCJD.[62] Possible factors attributing to an increase of CJD incidence is due to an aging population, population increase, clinician awareness, and more accurate diagnosis methods.[63] Since CJD symptoms are similar to other neurological conditions, it is also possible that CJD is mistaken for stroke, acute nephropathy, general dementia, and hyperparathyroidism.[63]
History
The disease was first described by German neurologist Hans Gerhard Creutzfeldt in 1920 and shortly afterward by Alfons Maria Jakob, giving it the name Creutzfeldt–Jakob. Some of the clinical findings described in their first papers do not match current criteria for Creutzfeldt–Jakob disease, and it has been speculated that at least two of the people in initial studies were suffering from a different ailment.[64] An early description of familial CJD stems from the German psychiatrist and neurologist Friedrich Meggendorfer (1880–1953).[65][66] A study published in 1997 counted more than 100 cases worldwide of transmissible CJD and new cases continued to appear at the time.[67]
The first report of suspected iatrogenic CJD was published in 1974. Animal experiments showed that corneas of infected animals could transmit CJD, and the causative agent spreads along visual pathways. A second case of CJD associated with a corneal transplant was reported without details. In 1977, CJD transmission caused by silver electrodes previously used in the brain of a person with CJD was first reported. Transmission occurred despite the decontamination of the electrodes with ethanol and formaldehyde. Retrospective studies identified four other cases likely of similar cause. The rate of transmission from a single contaminated instrument is unknown, although it is not 100%. In some cases, the exposure occurred weeks after the instruments were used on a person with CJD.[67] In the 1980s it was discovered that Lyodura, a dura mater transplant product was shown to transmit Creutzfeldt–Jakob disease from the donor to the recipient. This led to the product being banned in Canada but it was used in other countries such as Japan until 1993.[68]
A review article published in 1979 indicated that 25 dura mater cases had occurred by that date in Australia, Canada, Germany, Italy, Japan, New Zealand, Spain, the United Kingdom, and the United States.[67]
By 1985, a series of case reports in the United States showed that when injected, cadaver-extracted pituitary human growth hormone could transmit CJD to humans.[67]
In 1992, it was recognized that human gonadotropin administered by injection could also transmit CJD from person to person.[67]
Stanley B. Prusiner of the University of California, San Francisco (UCSF) was awarded the Nobel Prize in physiology or medicine in 1997 "for his discovery of Prions—a new biological principle of infection".[69] However, Yale University neuropathologist Laura Manuelidis has challenged the prion protein (PrP) explanation for the disease. In January 2007, she and her colleagues reported that they had found a virus-like particle in naturally and experimentally infected animals. "The high infectivity of comparable, isolated virus-like particles that show no intrinsic PrP by antibody labeling, combined with their loss of infectivity when nucleic acid–protein complexes are disrupted, make it likely that these 25-nm particles are the causal TSE virions".[70]
Australia
Australia has documented 10 cases of healthcare-acquired CJD (iatrogenic or ICJD). Five of the deaths resulted after the patients, who were in treatement either for infertility or short stature, were treated using contaminated pituitary extract hormone but no new cases have been noted since 1991. The other five deaths occurred due to dura grafting procedures that were performed during brain surgery, in which the covering of the brain is repaired. There have been no other ICJD deaths documented in Australia due to transmission during healthcare procedures.[71]
However, Carol Willesee, the ex-wife of Australian reporter Mike Willesee, died of the sporadic form of the disease in December 2006.[71][72]
New Zealand
A case was reported in 1989 in a 25-year-old man from New Zealand, who also received dura mater transplant.[67] Five New Zealanders have been confirmed to have died of the sporadic form of Creutzfeldt–Jakob disease (CJD) in 2012.[73]
United States
In 1988, there was a confirmed death from CJD of a person from Manchester, New Hampshire. Massachusetts General Hospital believed the person acquired the disease from a surgical instrument at a podiatrist's office. In September 2013, another person in Manchester was posthumously determined to have died of the disease. The person had undergone brain surgery at Catholic Medical Center three months before his death, and a surgical probe used in the procedure was subsequently reused in other operations. Public health officials identified thirteen people at three hospitals who may have been exposed to the disease through the contaminated probe, but said the risk of anyone's contracting CJD is "extremely low."[74][75][76] In January 2015, former speaker of the Utah House of Representatives Rebecca D. Lockhart died of the disease within a few weeks of diagnosis.[77] John Carroll, former editor of The Baltimore Sun and Los Angeles Times, died of CJD in Kentucky in June 2015, after having been diagnosed in January.[78] American actress Barbara Tarbuck (General Hospital, American Horror Story) died of the disease on December 26, 2016.[79]
Research
Diagnosis
- In 2010, a team from New York described detection of PrPSc in sheep's blood, even when initially present at only one part in one hundred billion (10−11) in sheep's brain tissue. The method combines amplification with a novel technology called surround optical fiber immunoassay (SOFIA) and some specific antibodies against PrPSc. The technique allowed improved detection and testing time for PrPSc.[80][81]
- In 2014, a human study showed a nasal brushing method that can accurately detect PrP in the olfactory epithelial cells of people with CJD.[82]
Treatment
- Pentosan polysulphate (PPS) was thought to slow the progression of the disease, and may have contributed to the longer than expected survival of the seven people studied.[83] The CJD Therapy Advisory Group to the UK Health Departments advises that data are not sufficient to support claims that pentosan polysulphate is an effective treatment and suggests that further research in animal models is appropriate.[84] A 2007 review of the treatment of 26 people with PPS finds no proof of efficacy because of the lack of accepted objective criteria.[85]
- Use of RNA interference to slow the progression of scrapie has been studied in mice. The RNA blocks production of the protein that the CJD process transforms into prions. This research is unlikely to lead to a human therapy for many years.[86]
- Both amphotericin B and doxorubicin have been investigated as treatments for CJD, but as yet there is no strong evidence that either drug is effective in stopping the disease. Further study has been taken with other medical drugs, but none are effective. However, anticonvulsants and anxiolytic agents, such as valproate or a benzodiazepine, may be administered to relieve associated symptoms.[12]
- Quinacrine, a medicine originally created for malaria, has been evaluated as a treatment for CJD. The efficacy of quinacrine was assessed in a rigorous clinical trial in the UK and the results were published in Lancet Neurology,[87] and concluded that quinacrine had no measurable effect on the clinical course of CJD.
- Astemizole, a medication approved for human use, has been found to have anti-prion activity and may lead to a treatment for Creutzfeldt–Jakob disease.[88]
- Use of Antisense oligonucleotides to slow progression of CJD are being investigated and have shown promising activity in mice models.[89]
References
- "Creutzfeldt-Jakob Disease, Classic (CJD)". CDC. 2 October 2018. Retrieved 21 November 2018.
- Mead S, Rudge P (April 2017). "CJD mimics and chameleons". Practical Neurology. 17 (2): 113–121. doi:10.1136/practneurol-2016-001571. PMC 5520355. PMID 28153848.
- Wells, John C. (2008), Longman Pronunciation Dictionary (3rd ed.), Longman, ISBN 9781405881180
- "Creutzfeldt–Jakob Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". NINDS. March 2003. Archived from the original on 4 July 2017. Retrieved 16 July 2017.
- "Creutzfeldt-Jakob Disease, Classic (CJD) | Prion Diseases | CDC". www.cdc.gov. 1 February 2019. Retrieved 17 June 2019.
Classic CJD is a human prion disease
- Manix, Marc; Kalakoti, Piyush; Henry, Miriam; Thakur, Jai; Menger, Richard; Guthikonda, Bharat; Nanda, Anil (2015-11-01). "Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy". Neurosurgical Focus. 39 (5): E2. doi:10.3171/2015.8.FOCUS15328. ISSN 1092-0684. PMID 26646926.
- "About CJD | Creutzfeldt–Jakob Disease, Classic (CJD) | Prion Disease". CDC. 11 February 2015. Archived from the original on 8 August 2017. Retrieved 16 July 2017.
- "Creutzfeldt–Jakob Disease, Classic (CJD) | Prion Diseases". CDC. 6 February 2015. Archived from the original on 18 July 2017. Retrieved 16 July 2017.
- "A 49-Year-Old Man With Forgetfulness and Gait Impairment". reference.medscape.com/viewarticle/881806_3. Archived from the original on 2017-07-06. Retrieved 2017-07-09.
- Murray ED, Buttner N, Price BH. (2012) Depression and Psychosis in Neurological Practice. In: Neurology in Clinical Practice, 6th Edition. Bradley WG, Daroff RB, Fenichel GM, Jankovic J (eds.) Butterworth Heinemann. April 12, 2012. ISBN 1437704344, 978-1437704341
- Brown P, Cathala F, Castaigne P, Gajdusek DC (November 1986). "Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases". Annals of Neurology. 20 (5): 597–602. doi:10.1002/ana.410200507. PMID 3539001. S2CID 7995631.
- Gambetti, Pierluigi. "Creutzfeldt–Jakob Disease (CJD)". The Merck Manuals: Online Medical Library. Archived from the original on 2011-06-04. Retrieved 2011-06-04.
- Atalay, Fatma Oz; Tolunay, Sahsine; Ozgun, Gonca; Bekar, Ahmet; Zarifoglu, Mehmet (2013). "Creutzfeldt-jakob disease: report of four cases and review of the literature". Turkish Journal of Pathology. doi:10.5146/tjpath.2013.01195. ISSN 1018-5615. PMID 24272930.
- Sattar HA (2017). Fundamentals of Pathology. p. 189. ISBN 9780983224631.
- Clarke AR, Jackson GS, Collinge J (February 2001). "The molecular biology of prion propagation". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 356 (1406): 185–95. doi:10.1098/rstb.2000.0764. PMC 1088424. PMID 11260799.
- Ridley RM, Baker HF, Crow TJ (February 1986). "Transmissible and non-transmissible neurodegenerative disease: similarities in age of onset and genetics in relation to aetiology". Psychological Medicine. 16 (1): 199–207. doi:10.1017/s0033291700002634. PMID 3961045.
- Will RG, Alperovitch A, Poser S, Pocchiari M, Hofman A, Mitrova E, de Silva R, D'Alessandro M, Delasnerie-Laupretre N, Zerr I, van Duijn C (June 1998). "Descriptive epidemiology of Creutzfeldt-Jakob disease in six European countries, 1993-1995. EU Collaborative Study Group for CJD". Annals of Neurology. 43 (6): 763–7. doi:10.1002/ana.410430611. PMID 9629846. S2CID 13562215.
- Armitage WJ, Tullo AB, Ironside JW (October 2009). "Risk of Creutzfeldt-Jakob disease transmission by ocular surgery and tissue transplantation". Eye. 23 (10): 1926–30. doi:10.1038/eye.2008.381. PMID 19136921.
- Esmonde T, Lueck CJ, Symon L, Duchen LW, Will RG (September 1993). "Creutzfeldt-Jakob disease and lyophilised dura mater grafts: report of two cases". Journal of Neurology, Neurosurgery, and Psychiatry. 56 (9): 999–1000. doi:10.1136/jnnp.56.9.999. PMC 489736. PMID 8410042.
- Bernoulli C, Siegfried J, Baumgartner G, Regli F, Rabinowicz T, Gajdusek DC, Gibbs CJ (February 1977). "Danger of accidental person-to-person transmission of Creutzfeldt-Jakob disease by surgery". Lancet. 1 (8009): 478–9. doi:10.1016/s0140-6736(77)91958-4. PMID 65575. S2CID 26804024.
- Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG, Ladogana A, Pocchiari M, Leschek EW, Schonberger LB (June 2012). "Iatrogenic Creutzfeldt-Jakob disease, final assessment". Emerging Infectious Diseases. 18 (6): 901–7. doi:10.3201/eid1806.120116. PMC 3358170. PMID 22607808.
- Brown P, Gibbs CJ, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC (May 1994). "Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease". Annals of Neurology. 35 (5): 513–29. doi:10.1002/ana.410350504. PMID 8179297. S2CID 22496358.
- New form of BSE resembles sporadic CJD
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF (October 1996). "Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD". Nature. 383 (6602): 685–90. Bibcode:1996Natur.383..685C. doi:10.1038/383685a0. PMID 8878476. S2CID 4355186.
- Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP (June 2006). "Kuru in the 21st century--an acquired human prion disease with very long incubation periods". Lancet. 367 (9528): 2068–74. doi:10.1016/s0140-6736(06)68930-7. PMID 16798390. S2CID 11506094.
- "Questions and Answers: Creutzfeldt–Jakob Disease Infection-Control Practices". Infection Control Practices/CJD (Creutzfeldt–Jakob Disease, Classic). Centers for Disease Control and Prevention. January 4, 2007. Archived from the original on 2007-10-17. Retrieved 2007-06-09.
- "WHO Infection Control Guidelines for Transmissible Spongiform Encephalopathies". World Health Organization: Communicable Disease Surveillance and Control. 26 March 1999. Archived from the original on 18 May 2007. Retrieved 2007-06-09.
- McDonnell G, Burke P (May 2003). "The challenge of prion decontamination". Clinical Infectious Diseases. 36 (9): 1152–4. doi:10.1086/374668. PMID 12715310.
- Solassol J, Pastore M, Crozet C, Perrier V, Lehmann S (September 2006). "A novel copper-hydrogen peroxide formulation for prion decontamination". The Journal of Infectious Diseases. 194 (6): 865–9. doi:10.1086/506947. PMID 16941355.
- Jackson GS, McKintosh E, Flechsig E, Prodromidou K, Hirsch P, Linehan J, Brandner S, Clarke AR, Weissmann C, Collinge J (March 2005). "An enzyme-detergent method for effective prion decontamination of surgical steel". The Journal of General Virology. 86 (Pt 3): 869–78. doi:10.1099/vir.0.80484-0. PMID 15722550.
- Sattar, Hussain A. (2011). Fundamentals of Pathology. Chicago: Pathoma LLC. p. 187.
- "Creutzfeldt–Jakob Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Retrieved 2018-01-13.
- Satoh J, Kurohara K, Yukitake M, Kuroda Y (1999). "The 14-3-3 protein detectable in the cerebrospinal fluid of patients with prion-unrelated neurological diseases is expressed constitutively in neurons and glial cells in culture". European Neurology. 41 (4): 216–25. doi:10.1159/000008054. PMID 10343153. S2CID 21795464.
- Geschwind MD, Martindale J, Miller D, DeArmond SJ, Uyehara-Lock J, Gaskin D, Kramer JH, Barbaro NM, Miller BL (June 2003). "Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease". Archives of Neurology. 60 (6): 813–6. doi:10.1001/archneur.60.6.813. PMID 12810484.
- Chitravas N, Jung RS, Kofskey DM, Blevins JE, Gambetti P, Leigh RJ, Cohen ML (September 2011). "Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease". Annals of Neurology. 70 (3): 437–44. doi:10.1002/ana.22454. PMC 3170496. PMID 21674591.
- Sano K, Satoh K, Atarashi R, Takashima H, Iwasaki Y, Yoshida M, Sanjo N, Murai H, Mizusawa H, Schmitz M, Zerr I, Kim YS, Nishida N (2013-01-25). "Early detection of abnormal prion protein in genetic human prion diseases now possible using real-time QUIC assay". PLOS ONE. 8 (1): e54915. Bibcode:2013PLoSO...854915S. doi:10.1371/journal.pone.0054915. PMC 3556051. PMID 23372790.
- Foutz A, Appleby BS, Hamlin C, Liu X, Yang S, Cohen Y, Chen W, Blevins J, Fausett C, Wang H, Gambetti P, Zhang S, Hughson A, Tatsuoka C, Schonberger LB, Cohen ML, Caughey B, Safar JG (January 2017). "Diagnostic and prognostic value of human prion detection in cerebrospinal fluid". Annals of Neurology. 81 (1): 79–92. doi:10.1002/ana.24833. PMC 5266667. PMID 27893164.
- Bongianni M, Orrù C, Groveman BR, Sacchetto L, Fiorini M, Tonoli G, Triva G, Capaldi S, Testi S, Ferrari S, Cagnin A, Ladogana A, Poleggi A, Colaizzo E, Tiple D, Vaianella L, Castriciano S, Marchioni D, Hughson AG, Imperiale D, Cattaruzza T, Fabrizi GM, Pocchiari M, Monaco S, Caughey B, Zanusso G (February 2017). "Diagnosis of Human Prion Disease Using Real-Time Quaking-Induced Conversion Testing of Olfactory Mucosa and Cerebrospinal Fluid Samples". JAMA Neurology. 74 (2): 155–162. doi:10.1001/jamaneurol.2016.4614. PMID 27942718. S2CID 40578363.
- Sanchez-Juan P, Green A, Ladogana A, Cuadrado-Corrales N, Sáanchez-Valle R, Mitrováa E, Stoeck K, Sklaviadis T, Kulczycki J, Hess K, Bodemer M, Slivarichová D, Saiz A, Calero M, Ingrosso L, Knight R, Janssens AC, van Duijn CM, Zerr I (August 2006). "CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease". Neurology. 67 (4): 637–43. doi:10.1212/01.wnl.0000230159.67128.00. PMID 16924018. S2CID 23306766.
- Tattum MH, Jones S, Pal S, Khalili-Shirazi A, Collinge J, Jackson GS (December 2010). "A highly sensitive immunoassay for the detection of prion-infected material in whole human blood without the use of proteinase K" (PDF). Transfusion (Submitted manuscript). 50 (12): 2619–27. doi:10.1111/j.1537-2995.2010.02731.x. PMID 20561299. S2CID 33032755.
- Fragoso DC, Gonçalves Filho AL, Pacheco FT, Barros BR, Aguiar Littig I, Nunes RH, Maia Júnior AC, da Rocha AJ (2017-01-01). "Imaging of Creutzfeldt-Jakob Disease: Imaging Patterns and Their Differential Diagnosis". Radiographics. 37 (1): 234–257. doi:10.1148/rg.2017160075. PMID 28076012.
- Finkenstaedt M, Szudra A, Zerr I, Poser S, Hise JH, Stoebner JM, Weber T (June 1996). "MR imaging of Creutzfeldt-Jakob disease". Radiology. 199 (3): 793–8. doi:10.1148/radiology.199.3.8638007. PMID 8638007.
- Ukisu R, Kushihashi T, Kitanosono T, Fujisawa H, Takenaka H, Ohgiya Y, Gokan T, Munechika H (February 2005). "Serial diffusion-weighted MRI of Creutzfeldt-Jakob disease". AJR. American Journal of Roentgenology. 184 (2): 560–6. doi:10.2214/ajr.184.2.01840560. PMID 15671380.
- Young GS, Geschwind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S, Lu Y, Wong S, Liu H, Miller BL, Dillon WP (June–July 2005). "Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis". AJNR. American Journal of Neuroradiology. 26 (6): 1551–62. PMID 15956529. Archived from the original on 2008-09-07.
- Abdulmassih R, Min Z (March 2016). "An ominous radiographic feature: cortical ribbon sign". Internal and Emergency Medicine. 11 (2): 281–3. doi:10.1007/s11739-015-1287-4. PMID 26238299.
- Tschampa HJ, Mürtz P, Flacke S, Paus S, Schild HH, Urbach H (May 2003). "Thalamic involvement in sporadic Creutzfeldt-Jakob disease: a diffusion-weighted MR imaging study". AJNR. American Journal of Neuroradiology. 24 (5): 908–15. PMID 12748093. Archived from the original on 10 October 2008.
- Sternberg's Diagnostic Surgical Pathology, 5th edition.
- Liberski, P.P. (2004). "Spongiform change – an electron microscopic view". Folia Neuropathologica. 42, suppl B: 59–70. PMID 16903142.
- "Pathology of Degenerative CNS Diseases". library.med.utah.edu.
- Budka H, Will RG (12 November 2015). "The end of the BSE saga: do we still need surveillance for human prion diseases?" (PDF). Swiss Medical Weekly. 145: w14212. doi:10.4414/smw.2015.14212. PMID 26715203.
- "who.int: "Fact sheets no 180: Variant Creutzfeldt–Jakob disease" Feb 2012 ed". Archived from the original on February 4, 2016.
- Bonda DJ, Manjila S, Mehndiratta P, Khan F, Miller BR, Onwuzulike K, Puoti G, Cohen ML, Schonberger LB, Cali I (July 2016). "Human prion diseases: surgical lessons learned from iatrogenic prion transmission". Neurosurgical Focus. 41 (1): E10. doi:10.3171/2016.5.FOCUS15126. PMC 5082740. PMID 27364252.
- "Creutzfeldt-Jakob Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". NINDS. Retrieved 21 November 2018.
- Belay ED, Schonberger LB (December 2002). "Variant Creutzfeldt-Jakob disease and bovine spongiform encephalopathy". Clinics in Laboratory Medicine. 22 (4): 849–62, v–vi. doi:10.1016/S0272-2712(02)00024-0. PMID 12489284.
- Manix M, Kalakoti P, Henry M, Thakur J, Menger R, Guthikonda B, Nanda A (November 2015). "Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy". Neurosurgical Focus. 39 (5): E2. doi:10.3171/2015.8.FOCUS15328. PMID 26646926.
- "Treatment". nhs.uk. 2017-10-23. Retrieved 2018-04-17.
- Ng MC, Westover MB, Cole AJ (2014-02-07). "Treating seizures in Creutzfeldt-Jakob disease". Epilepsy & Behavior Case Reports. 2: 75–9. doi:10.1016/j.ebcr.2014.01.004. PMC 4308028. PMID 25667875.
- Mizutani T, Okumura A, Oda M, Shiraki H (February 1981). "Panencephalopathic type of Creutzfeldt-Jakob disease: primary involvement of the cerebral white matter". Journal of Neurology, Neurosurgery, and Psychiatry. 44 (2): 103–15. doi:10.1136/jnnp.44.2.103. PMC 490840. PMID 7012278.
- "CJD (Creutzfeldt–Jakob Disease, Classic)". Centers for Disease Control and Prevention. 2008-02-26. Archived from the original on 2009-05-06. Retrieved 2009-06-20.
- "vCJD (Variant Creutzfeldt–Jakob Disease)". Centers for Disease Control and Prevention. 2007-01-04. Archived from the original on 2009-05-07. Retrieved 2009-06-20.
- "Occurrence and Transmission | Creutzfeldt-Jakob Disease, Classic (CJD) | Prion Disease | CDC". www.cdc.gov. 2019-05-08. Retrieved 2020-11-04.
- Bosque, Patrick; Tyler, Kenneth (2020). Prions and Prion Disease of the Central Nervous System (Transmissible Neurodegenerative Diseases). www.clinicalkey.com: Elsevier, Inc. pp. 2288–2300.
- Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. 2020 Jan;20(1):e2-e10. doi: 10.1016/S1473-3099(19)30615-2. PMID: 31876504.
- Ironside JW (1996). "Neuropathological diagnosis of human prion disease; morphological studies". In Baker HF, Ridley RM (eds.). Prion Diseases. 3. pp. 35–57. doi:10.1385/0896033422. ISBN 978-0-89603-342-9.
- Meggendorfer F (1930). "Klinische und genealogische Beobachtungen bei einem Fall von spastischer Pseudokosklerose Jakobs". Zeitschrift für die Gesamte Neurologie und Psychiatrie. 128 (1–4): 337–41. doi:10.1007/bf02864269.
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003). "Sporadic and familial CJD: classification and characterisation". British Medical Bulletin. 66: 213–39. doi:10.1093/bmb/66.1.213. PMID 14522861.
- Ricketts MN, Cashman NR, Stratton EE, ElSaadany S (1997). "Is Creutzfeldt-Jakob disease transmitted in blood?". Emerging Infectious Diseases. 3 (2): 155–63. doi:10.3201/eid0302.970208. PMC 2627622. PMID 9204296.
- Ae R, Hamaguchi T, Nakamura Y, Yamada M, Tsukamoto T, Mizusawa H, Belay ED, Schonberger LB (March 2018). "Update: Dura Mater Graft-Associated Creutzfeldt-Jakob Disease - Japan, 1975-2017". MMWR. Morbidity and Mortality Weekly Report. 67 (9): 274–278. doi:10.15585/mmwr.mm6709a3. PMC 5844283. PMID 29518068.
- "The Nobel Prize in Physiology or Medicine 1997: Stanley B. Prusiner". NobelPrize.org. Archived from the original on 2011-02-20. Retrieved 2011-02-21.
- Manuelidis L, Yu ZX, Barquero N, Banquero N, Mullins B (February 2007). "Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles". Proceedings of the National Academy of Sciences of the United States of America. 104 (6): 1965–70. Bibcode:2007PNAS..104.1965M. doi:10.1073/pnas.0610999104. PMC 1794316. PMID 17267596.
- Creutzfeldt–Jakob Disease (CJD) – the facts. Infectious Diseases Epidemiology & Surveillance – Department of Health, Victoria, Australia Archived 2015-06-28 at the Wayback Machine
- Reporter, Natasha Wallace Health (15 April 2008). "Hospital 'failed' to diagnose actress". The Sydney Morning Herald.
- "Mad cow link in hunter's death". Stuff.
- "Autopsy confirms rare brain disease in NH patient". MyFoxBoston. 2013-09-20. Archived from the original on 21 September 2013. Retrieved 20 September 2013.
- "NH Patient Likely Died of Rare Brain Disease". AP. Archived from the original on 8 September 2013. Retrieved 5 September 2013.
- Kowalczyk, Liz. "5 patients at Cape hospital at risk for rare brain disease". Boston Globe. Archived from the original on 9 September 2013. Retrieved 25 November 2013.
- "Officials: Lockhart, 'Utah's Iron Lady' died from Creutzfeldt-Jakob disease". Archived from the original on 2015-01-20. Retrieved 2015-01-18.
- Schudel, Matt (June 14, 2015). "John S. Carroll, acclaimed newspaper editor in Baltimore and L.A., dies at 73". The Washington Post. Archived from the original on September 19, 2016.
- "Barbara Tarbuck, 'General Hospital' and 'American Horror Story' Actress, Dies at 74". 2016-12-30. Archived from the original on 2016-12-30. Retrieved 2016-12-30.
- "Detecting Prions in Blood" (PDF). Microbiology Today: 195. August 2010. Archived from the original (PDF) on 31 March 2012. Retrieved 2011-08-21.
- Rubenstein R, Chang B, Gray P, Piltch M, Bulgin MS, Sorensen-Melson S, Miller MW (July 2010). "A novel method for preclinical detection of PrPSc in blood". The Journal of General Virology. 91 (Pt 7): 1883–92. doi:10.1099/vir.0.020164-0. PMID 20357038.
- Orrú CD, Bongianni M, Tonoli G, Ferrari S, Hughson AG, Groveman BR, Fiorini M, Pocchiari M, Monaco S, Caughey B, Zanusso G (August 2014). "A test for Creutzfeldt-Jakob disease using nasal brushings". The New England Journal of Medicine. 371 (6): 519–29. doi:10.1056/NEJMoa1315200. PMC 4186748. PMID 25099576.
- Bone, Ian (12 July 2006). "Intraventricular Pentosan Polysulphate in Human Prion Diseases: A study of Experience in the United Kingdom". Medical Research Council. Archived from the original on 3 November 2012. Retrieved 2012-11-09.
- "Use of Pentosan Polysulphate in the treatment of, or prevention of, vCJD". Department of Health:CJD Therapy Advisory Group. Archived from the original on 2007-07-05. Retrieved 2007-10-30.
- Rainov NG, Tsuboi Y, Krolak-Salmon P, Vighetto A, Doh-Ura K (May 2007). "Experimental treatments for human transmissible spongiform encephalopathies: is there a role for pentosan polysulfate?". Expert Opinion on Biological Therapy. 7 (5): 713–26. doi:10.1517/14712598.7.5.713. PMID 17477808. S2CID 12725001.
- Pfeifer A, Eigenbrod S, Al-Khadra S, Hofmann A, Mitteregger G, Moser M, Bertsch U, Kretzschmar H (December 2006). "Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice". The Journal of Clinical Investigation. 116 (12): 3204–10. doi:10.1172/JCI29236. PMC 1679709. PMID 17143329. Lay summary – BBC News (2006-12-04).
- Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, Thomas D, Walker S, Webb T, Wroe S, Darbyshire J (April 2009). "Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial". The Lancet. Neurology. 8 (4): 334–44. doi:10.1016/S1474-4422(09)70049-3. PMC 2660392. PMID 19278902.
- Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, Chase P, Fallahi M, Hodder P, Weissmann C, Lasmézas CI (April 2013). "Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents". Proceedings of the National Academy of Sciences of the United States of America. 110 (17): 7044–9. Bibcode:2013PNAS..110.7044K. doi:10.1073/pnas.1303510110. PMC 3637718. PMID 23576755.
- "Prion disease in mice treated successfully with antisense oligonucleotides". News-Medical.net. 2019-08-01. Retrieved 2019-08-09.
External links
| Classification | |
|---|---|
| External resources |