Cyclopentadienyliron dicarbonyl dimer
Cyclopentadienyliron dicarbonyl dimer is an organometallic compound with the formula [(η5-C5H5)Fe(CO)2]2, often abbreviated to Cp2Fe2(CO)4, [CpFe(CO)2]2 or even Fp2, with the colloquial name "fip dimer". It is a dark reddish-purple crystalline solid, which is readily soluble in moderately polar organic solvents such as chloroform and pyridine, but less soluble in carbon tetrachloride and carbon disulfide. Cp2Fe2(CO)4 is insoluble in but stable toward water. Cp2Fe2(CO)4 is reasonably stable to storage under air and serves as a convenient starting material for accessing other Fp (CpFe(CO)2) derivatives (described below).[1]
 | |
 | |
| Names | |
|---|---|
| IUPAC name
Di-μ-carbonyldicarbonylbis(η5-cyclopenta-2,4-dien-1-yl)diiron | |
| Other names
Bis(cyclopentadienyl)tetracarbonyl-diiron, Di(cyclopentadienyl)tetracarbonyl-diiron, Bis(dicarbonylcyclopentadienyliron) | |
| Identifiers | |
3D model (JSmol) |
|
| ChemSpider | |
| ECHA InfoCard | 100.032.057 |
| EC Number |
|
PubChem CID |
|
| |
| |
| Properties | |
| C14H10Fe2O4 | |
| Molar mass | 353.925 g/mol |
| Appearance | Dark purple crystals |
| Density | 1.77 g/cm3, solid |
| Melting point | 194 °C (381 °F; 467 K) |
| Boiling point | decomposition |
| insoluble | |
| Solubility in other solvents | benzene, THF, chlorocarbons |
| Structure | |
| distorted octahedral | |
| 3.1 D (benzene solution) | |
| Hazards | |
| Main hazards | CO source |
| GHS pictograms |    |
| GHS Signal word | Danger |
| H228, H302, H331, H330 | |
| Related compounds | |
Related compounds |
Fe(C5H5)2 Fe(CO)5 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
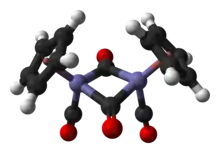
Structure
In solution, Cp2Fe2(CO)4 can be considered a dimeric half-sandwich complex. It exists in three isomeric forms: cis, trans, and an unbridged, open form. These isomeric forms are distinguished by the position of the ligands. The cis and trans isomers differ in the relative position of C5H5 (Cp) ligands. The cis and trans isomers have the formulation [(η5-C5H5)Fe(CO)(μ-CO)]2, that is, two CO ligands are terminal whereas the other two CO ligands bridge between the iron atoms. The cis and trans isomers interconvert via the open isomer, which has no bridging ligands between iron atoms. Instead, it is formulated as (η5-C5H5)(OC)2Fe−Fe(CO)2(η5-C5H5) — the metals are held together by an iron–iron bond. At equilibrium, the cis and trans isomers are predominant.

In addition, the terminal and bridging carbonyls are known to undergo exchange: the trans isomer can undergo bridging–terminal CO ligand exchange through the open isomer, or through a twisting motion without going through the open form. In contrast, the bridging and terminal CO ligands of the cis isomer can only exchange via the open isomer.[2]
In solution, the cis, trans, and open isomers interconvert rapidly at room temperature, making the molecular structure fluxional. The fluxional process for cyclopentadienyliron dicarbonyl dimer is faster than the NMR time scale, so that only an averaged, single Cp signal is observed in the 1H NMR spectrum at 25 °C. Likewise, the 13C NMR spectrum exhibits one sharp CO signal above −10 °C, while the Cp signal sharpens to one peak above 60 °C. NMR studies indicate that the cis isomer is slightly more abundant than the trans isomer at room temperature, while the amount of the open form is small.[2] The fluxional process is not fast enough to produce averaging in the IR spectrum. Thus, three absorptions are seen for each isomer. The bridging CO ligands appear at around 1780 cm−1 whereas the terminal CO ligands are observed at around 1980 cm−1.[3]
The solid-state molecular structure of both cis and trans isomers have been analyzed by X-ray and neutron diffraction. The Fe–Fe separation and the Fe–C bond lengths are the same in the Fe2C2 rhomboids, an exactly planar Fe2C2 four-membered ring in the trans isomer versus a folded rhomboid in cis with an angle of 164°, and significant distortions in the Cp ring of the trans isomer reflecting different Cp orbital populations.[4] Although older textbooks show the two iron atoms bonded to each other, theoretical analyses indicate the absence of a direct Fe–Fe bond. This view is consistent with computations and X-ray crystallographic data that indicate a lack of significant electron density between the iron atoms.[5] However, Labinger offers a dissenting view, based primarily on chemical reactivity and spectroscopic data, arguing that electron density is not necessarily the best indication of the presence of a chemical bond. Moreover, without an Fe–Fe bond, the bridging carbonyls must be formally treated as an μ-X2 ligand and μ-L ligand in order for the iron centers to satisfy the 18-electron rule. This formalism is argued to give misleading implications with respect to the chemical and spectroscopic behavior of the carbonyl groups.[6]
The averaged structure of these isomers of Cp2Fe2(CO)4 results in a dipole moment of 3.1 D in benzene.[7]
Synthesis
Cp2Fe2(CO)4 was first prepared in 1955 at Harvard by Geoffrey Wilkinson using the same method employed today: the reaction of iron pentacarbonyl and dicyclopentadiene.[6][8]
- 2 Fe(CO)5 + C10H12 → (η5-C5H5)2Fe2(CO)4 + 6 CO + H2
In this preparation, dicyclopentadiene cracks to give cyclopentadiene, which reacts with Fe(CO)5 with loss of CO. Thereafter, the pathways for the photochemical and thermal routes differ subtly but both entail formation of a hydride intermediate.[4] The method is used in the teaching laboratory.[3]
Reactions
Although of no major commercial value, Fp2 is a workhorse in organometallic chemistry because it is inexpensive and FpX derivatives are rugged (X = halide, organyl).
"Fp−" (FpNa and FpK)
Reductive cleavage of [CpFe(CO)2]2 (formally an iron(I) complex) produces alkali metal derivatives formally derived from the cyclopentadienyliron dicarbonyl anion, [CpFe(CO)2]− or called Fp− (formally iron(0)), which are assumed to exist as a tight ion pair. A typical reductant is sodium metal or sodium amalgam;[9] NaK alloy, potassium graphite (KC8), and alkali metal trialkylborohydrides have been used. [CpFe(CO)2]Na is a widely studied reagent since it is readily alkylated, acylated, or metalated by treatment with an appropriate electrophile.[10] It is an excellent SN2 nucleophile, being one to two orders of magnitude more nucleophilic than thiophenolate, PhS– when reacted with primary and secondary alkyl bromides.[11]
- [CpFe(CO)2]2 + 2 Na → 2 CpFe(CO)2Na
- [CpFe(CO)2]2 + 2 KBH(C2H5)3 → 2 CpFe(CO)2K + H2 + 2 B(C2H5)3
Treatment of NaFp with an alkyl halide (RX, X = Br, I) produces FeR(η5-C5H5)(CO)2
- CpFe(CO)2K + CH3I → CpFe(CO)2CH3 + KI
Fp2 can also be cleaved with alkali metals[12] and by electrochemical reduction.[13][14]
FpX (X = Cl, Br, I)
Halogens oxidatively cleave [CpFe(CO)2]2 to give the Fe(II) species FpX (X = Cl, Br, I):
- [CpFe(CO)2]2 + X2 → 2 CpFe(CO)2X
One example is cyclopentadienyliron dicarbonyl iodide.
Fp(η2-alkene)+, Fp(η2-alkyne)+ and other "Fp+"
In the presence of halide anion acceptors such as AlBr3 or AgBF4, FpX compounds (X = halide) react with alkenes, alkynes, or neutral labile ligands (such as ethers and nitriles) to afford Fp+ complexes.[15] In another approach, salts of [Fp(isobutene)]+ are readily obtained by reaction of NaFp with methallyl chloride followed by protonolysis. This complex is a convenient and general precursor to other cationic Fp–alkene and Fp–alkyne complexes.[16] The exchange process is facilitated by the loss of gaseous and bulky isobutene.[17] Generally, less substituted alkenes bind more strongly and can displace more hindered alkene ligands. Alkene and alkyne complexes can also be prepared by heating a cationic ether or aqua complex, for example [Fp(thf)]+
BF−
4, with the alkene or alkyne.[18] [FpL]+
BF−
4 complexes can also be prepared by treatment of FpMe with HBF4·Et2O in CH2Cl2 at −78 °C, followed by addition of L.[19]
cation.png.webp)
Alkene–Fp complexes can also be prepared from Fp anion indirectly. Thus, hydride abstraction from Fp–alkyl compounds using triphenylmethyl hexafluorophosphate affords [Fp(α-alkene)]+ complexes.
- FpNa + RCH2CH2I → FpCH2CH2R + NaI
- FpCH2CH2R + Ph3CPF6 → [Fp(CH
2=CHR)+
]PF−
6 + Ph3CH
Reaction of NaFp with an epoxide followed by acid-promoted dehydration also affords alkene complexes. Fp(alkene)+ are stable with respect to bromination, hydrogenation, and acetoxymercuration, but the alkene is easily released with sodium iodide in acetone or by warming with acetonitrile.[20]
exchange.png.webp)
The alkene ligand in these cations is activated toward attack by nucleophiles, opening the way to a number of carbon–carbon bond-forming reactions. Nucleophilic additions usually occur at the more substituted carbon. This regiochemistry is attributed to the greater positive charge density at this position. The regiocontrol is often modest. The addition of the nucleophile is completely stereoselective, occurring anti to the Fp group. Analogous Fp(alkyne)+ complexes are also reported to undergo nucleophilic addition reactions by various carbon, nitrogen, and oxygen nucleophiles.[21]

Fp(alkene)+ and Fp(alkyne)+ π-complexes are also quite acidic at the allylic and propargylic positions, respectively, and can be quantitatively deprotonated with amine bases like Et3N to give neutral Fp–allyl and Fp–allenyl σ-complexes:[16]
- Fp(H
2C=CHCH
2CH
3)+
BF−
4 + Et3N → FpCH2CH=CHCH3 + Et
3NH+
BF−
4 - FpCH2CH=CHCH3 + E+
BF−
4 → Fp(H
2C=CHCH(E)CH
3)+
BF−
4
Fp–allyl and Fp–allenyl react with cationic electrophiles E (such as Me3O+, carbocations, oxocarbenium ions) to generate allylic and propargylic functionalization products, respectively.[16] The related complex [Cp*Fe(CO)2(thf)]+[BF4]− has been shown to catalyze propargylic and allylic C−H functionalization by leveraging the deprotonation and electrophilic functionalization processes described above.[22]
Fp-based cyclopropanation reagents
Fp-based reagents have been developed for cyclopropanations.[23] The key reagent is prepared from FpNa with a thioether and methyl iodide, and has a good shelf-life, in contrast to typical Simmons-Smith intermediates and diazoalkanes.
- FpNa + ClCH2SCH3 → FpCH2SCH3 + NaCl
- FpCH2SCH3 + CH3I + NaBF4 → FpCH2S(CH3)2]BF4 + NaI
Use of [FpCH2S(CH3)2]BF4 does not require specialized conditions.
- Fp(CH
2S+
(CH
3)
2)BF−
4 + (Ph)2C=CH2 → 1,1-diphenylcyclopropane + …
Iron(III) chloride is added to destroy any byproduct.
Precursors to Fp=CH+
2, like FpCH2OMe which is converted to the iron carbene upon protonation, have also been used as cyclopropanation reagents.[24]
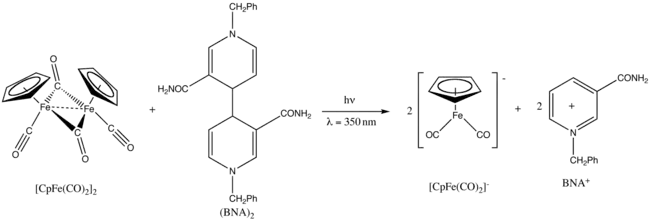
Photochemical reaction
Fp2 exhibits photochemistry.[25] Upon UV irradiation at 350 nm, it is reduced by 1-benzyl-1,4-dihydronicotinamide dimer, (BNA)2.[26]
References
- Kelly, William J. (2001). "Bis(dicarbonylcyclopentadienyliron)". Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rb139. ISBN 0471936235.
- Harris, Daniel C.; Rosenberg, Edward; Roberts, John D. (1974). "Carbon-13 nuclear magnetic resonance spectra and mechanism of bridge–terminal carbonyl exchange in di-µ-carbonyl-bis[carbonyl(η-cyclopentadienyl)iron](Fe–Fe) [{(η-C5H5)Fe(CO)2}2]; cd-di-µ-carbonyl-f-carbonyl-ae-di(η-cyclopentadienyl)-b-(triethyl-phosphite)di-iron(Fe–Fe) [(η-C5H5)2Fe2(CO)3P(OEt)3], and some related complexes". Journal of the Chemical Society: Dalton Transactions (22): 2398–2403. doi:10.1039/DT9740002398. ISSN 0300-9246.
- Girolami, G.; Rauchfuss, T.; Angelici, R. (1999). Synthesis and Technique in Inorganic Chemistry (3rd ed.). Sausalito, CA: University Science Books. pp. 171–180. ISBN 978-0-935702-48-4.
- Wilkinson, G., ed. (1982). Comprehensive Organometallic Chemistry. 4. New York: Pergamon Press. pp. 513–613. ISBN 978-0-08-025269-8.
- Green, Jennifer C.; Green, Malcolm L. H.; Parkin, Gerard (2012). "The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds". Chemical Communications. 2012 (94): 11481–11503. doi:10.1039/c2cc35304k. PMID 23047247.
- Labinger, Jay A. (2015). "Does cyclopentadienyl iron dicarbonyl dimer have a metal–metal bond? Who's asking?". Inorganica Chimica Acta. Metal–Metal Bonded Compounds and Metal Clusters. 424: 14–19. doi:10.1016/j.ica.2014.04.022. ISSN 0020-1693.
- Cotton, F. Albert; Yagupsky, G. (1967). "Tautomeric changes in metal carbonyls. I. π-Cyclopentadienyliron dicarbonyl dimer and π-cyclopentadienyl-ruthenum dicarbonyl dimer". Inorganic Chemistry. 6 (1): 15–20. doi:10.1021/ic50047a005. ISSN 0020-1669.
- Piper, T. S.; Cotton, F. A.; Wilkinson, G. (1955). "Cyclopentadienyl–carbon monoxide and related compounds of some transitional metals". Journal of Inorganic and Nuclear Chemistry. 1 (3): 165–174. doi:10.1016/0022-1902(55)80053-X.
- Chang, T. C. T.; Rosenblum, M.; Simms, N. (1988). "Vinylation of Enolates with a Vinyl Cation Equivalent: trans-3-Methyl-2-Vinylcyclohexanone". Organic Syntheses. 66: 95.; Collective Volume, 8, p. 479
- King, B. (1970). "Applications of Metal Carbonyl Anions in the Synthesis of Unusual Organometallic Compounds". Accounts of Chemical Research. 3 (12): 417–427. doi:10.1021/ar50036a004.
- Dessy, Raymond E.; Pohl, Rudolph L.; King, R. Bruce (1966-11-01). "Organometallic Electrochemistry. VII.1 The Nucleophilicities of Metallic and Metalloidal Anions Derived from Metals of Groups IV, V, VI, VII, and VIII". Journal of the American Chemical Society. 88 (22): 5121–5124. doi:10.1021/ja00974a015. ISSN 0002-7863.
- Ellis, J. E.; Flom, E. A. (1975). "The Chemistry of Metal Carbonyl Anions: III. Sodium-Potassium Alloy: An Efficient Reagent for the Production of Metal Carbonyl Anions". Journal of Organometallic Chemistry. 99 (2): 263–268. doi:10.1016/S0022-328X(00)88455-7.
- Dessy, R. E.; King, R. B.; Waldrop, M. (1966). "Organometallic Electrochemistry. V. The Transition Series". Journal of the American Chemical Society. 88 (22): 5112–5117. doi:10.1021/ja00974a013.
- Dessy, R. E.; Weissman, P. M.; Pohl, R. L. (1966). "Organometallic Electrochemistry. VI. Electrochemical Scission of Metal–Metal Bonds". Journal of the American Chemical Society. 88 (22): 5117–5121. doi:10.1021/ja00974a014.
- Silver, J. (1993). Chemistry of Iron. Dordrecht: Springer Netherlands. ISBN 9789401121408. OCLC 840309324.
- Cutler, A.; Ehnholt, D.; Lennon, P.; Nicholas, K.; Marten, David F.; Madhavarao, M.; Raghu, S.; Rosan, A.; Rosenblum, M. (1975). "Chemistry of dicarbonyl .eta.5-cyclopentadienyliron complexes. General syntheses of monosubstituted η2-olefin complexes and of 1-substituted η1-allyl complexes. Conformational effects on the course of deprotonation of (η2-olefin) cations". Journal of the American Chemical Society. 97 (11): 3149–3157. doi:10.1021/ja00844a038. ISSN 0002-7863.
- Turnbull, Mark M. (2001). "Dicarbonyl(cyclopentadienyl)(isobutene)iron Tetrafluoroborate". Encyclopedia of Reagents for Organic Synthesis. eEROS. doi:10.1002/047084289X.rd080. ISBN 0471936235.
- Schriver, D. F.; Bruce, M. I.; Wilkinson, G. (1995). Iron, Ruthenium and Osmium. Kidlington: Elsevier Science. ISBN 978-0-08-096396-9. OCLC 953660855.
- Redlich, Mark D.; Mayer, Michael F.; Hossain, M. Mahmun (2003). "Iron Lewis Acid [(η5-C5H5)Fe(CO)2(THF)]+ Catalyzed Organic Reactions". Aldrichimica Acta. 36: 3–13.
- Pearson, A. J. (1994). Iron Compounds in Organic Synthesis. San Diego, CA: Academic Press. pp. 22–35. ISBN 978-0-12-548270-7.
- Akita, Munetaka; Kakuta, Satoshi; Sugimoto, Shuichiro; Terada, Masako; Tanaka, Masako; Moro-oka, Yoshihiko (2001). "Nucleophilic Addition to the η2-Alkyne Ligand in [CpFe(CO)2(η2-R−C⋮C−R)]+. Dependence of the Alkenyl Product Stereochemistry on the Basicity of the Nucleophile". Organometallics. 20 (13): 2736–2750. doi:10.1021/om010095t. ISSN 0276-7333.
- Wang, Yidong; Zhu, Jin; Durham, Austin C.; Lindberg, Haley; Wang, Yi-Ming (2019). "α-C–H Functionalization of π-Bonds Using Iron Complexes: Catalytic Hydroxyalkylation of Alkynes and Alkenes". Journal of the American Chemical Society. 141 (50): 19594–19599. doi:10.1021/jacs.9b11716. ISSN 0002-7863. PMID 31791121.
- Mattson, M. N.; O'Connor, E. J.; Helquist, P. (1992). "Cyclopropanation Using an Iron-Containing Methylene Transfer Reagent: 1,1-Diphenylcyclopropane". Organic Syntheses. 70: 177.; Collective Volume, 9, p. 372
- Johnson, M. D. (1982), "Mononuclear Iron Compounds with η1-Hydrocarbon Ligands", Comprehensive Organometallic Chemistry, Elsevier, pp. 331–376, doi:10.1016/b978-008046518-0.00049-0, ISBN 978-0-08-046518-0, retrieved 2019-12-11
- Wrighton, M. (1974). "Photochemistry of Metal Carbonyls". Chemical Reviews. 74 (4): 401–430. doi:10.1021/cr60290a001.
- Fukuzumi, S.; Ohkubo, K.; Fujitsuka, M.; Ito, O.; Teichmann, M. C.; Maisonhaute, E.; Amatore, C. (2001). "Photochemical Generation of Cyclopentadienyliron Dicarbonyl Anion by a Nicotinamide Adenine Dinucleotide Dimer Analogue". Inorganic Chemistry. 40 (6): 1213–1219. doi:10.1021/ic0009627. PMID 11300821.