De novo gene birth
De novo gene birth is the process by which new genes evolve from DNA sequences that were ancestrally non-genic.[3] De novo genes represent a subset of novel genes, and may be protein-coding or instead act as RNA genes.[4] The processes that govern de novo gene birth are not well understood, although several models exist that describe possible mechanisms by which de novo gene birth may occur.
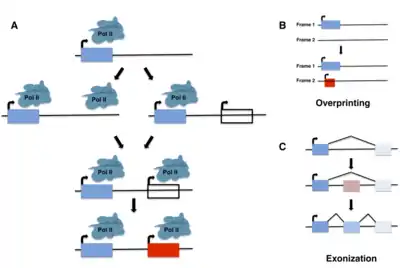
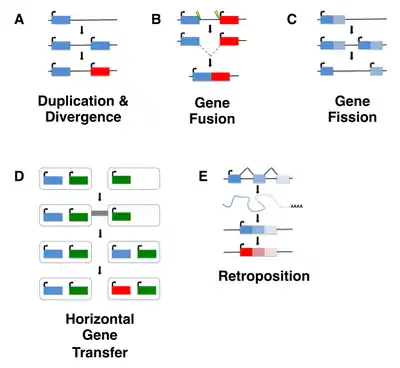
Although de novo gene birth may have occurred at any point in an organism's evolutionary history, ancient de novo gene birth events are difficult to detect. Most studies of de novo genes to date have thus focused on young genes, typically taxonomically restricted genes (TRGs) that are present in a single species or lineage, including so-called orphan genes, defined as genes that lack any identifiable homolog. It is important to note, however, that not all orphan genes arise de novo, and instead may emerge through fairly well characterized mechanisms such as gene duplication (including retroposition) or horizontal gene transfer followed by sequence divergence, or by gene fission/fusion.[5][6]
Although de novo gene birth was once viewed as a highly unlikely occurrence,[7] several unequivocal examples have now been described,[8] and some researchers speculate that de novo gene birth could play a major role in evolutionary innovation.[9][10]
History
As early as the 1930s, J. B. S. Haldane and others suggested that copies of existing genes may lead to new genes with novel functions.[6] In 1970, Susumu Ohno published the seminal text Evolution by Gene Duplication.[11] For some time subsequently, the consensus view was that virtually all genes were derived from ancestral genes,[12] with François Jacob famously remarking in a 1977 essay that "the probability that a functional protein would appear de novo by random association of amino acids is practically zero."[7]
In the same year, however, Pierre-Paul Grassé coined the term "overprinting" to describe the emergence of genes through the expression of alternative open reading frames (ORFs) that overlap preexisting genes.[13] These new ORFs may be out of frame with or antisense to the preexisting gene. They may also be in frame with the existing ORF, creating a truncated version of the original gene, or represent 3’ extensions of an existing ORF into a nearby ORF. The first two types of overprinting may be thought of as a particular subtype of de novo gene birth; although overlapping with a previously coding region of the genome, the primary amino-acid sequence of the new protein is entirely novel and derived from a frame that did not previously contain a gene. The first examples of this phenomenon in bacteriophages were reported in a series of studies from 1976 to 1978,[14][15][16] and since then numerous other examples have been identified in viruses, bacteria, and several eukaryotic species.[17][18][19][20][21][22]
The phenomenon of exonization also represents a special case of de novo gene birth, in which, for example, often-repetitive intronic sequences acquire splice sites through mutation, leading to de novo exons. This was first described in 1994 in the context of Alu sequences found in the coding regions of primate mRNAs.[23] Interestingly, such de novo exons are frequently found in minor splice variants, which may allow the evolutionary “testing” of novel sequences while retaining the functionality of the major splice variant(s).[24]
Still, it was thought by some that most or all eukaryotic proteins were constructed from a constrained pool of “starter type” exons.[25] Using the sequence data available at the time, a 1991 review estimated the number of unique, ancestral eukaryotic exons to be < 60,000,[25] while in 1992 a piece was published estimating that the vast majority of proteins belonged to no more than 1,000 families.[26] Around the same time, however, the sequence of chromosome III of the budding yeast Saccharomyces cerevisiae was released,[27] representing the first time an entire chromosome from any eukaryotic organism had been sequenced. Sequencing of the entire yeast nuclear genome was then completed by early 1996 through a massive, collaborative international effort.[28] In his review of the yeast genome project, Bernard Dujon noted that the unexpected abundance of genes lacking any known homologs was perhaps the most striking finding of the entire project.[28]
In 2006 and 2007, a series of studies provided arguably the first documented examples of de novo gene birth that did not involve overprinting.[29][30][31] An analysis of the accessory gland transcriptomes of Drosophila yakuba and Drosophila erecta first identified 20 putative lineage-restricted genes that appeared unlikely to have resulted from gene duplication.[31] Levine and colleagues then confirmed the de novo origination of five candidate genes specific to Drosophila melanogaster and/or the closely related Drosophila simulans through a rigorous pipeline that combined bioinformatic and experimental techniques.[30] These genes were identified by combining BLAST search-based and synteny-based approaches (see below), which demonstrated the absence of the genes in closely related species.[30]
Despite their recent evolution, all five genes appear fixed in D. melanogaster, and the presence of paralogous non-coding sequences that are absent in close relatives suggests that four of the five genes may have arisen through a recent intrachromosomal duplication event.[30] Interestingly, all five were preferentially expressed in the testes of male flies[30] (see below). The three genes for which complete ORFs exist in both D. melanogaster and D. simulans showed evidence of rapid evolution and positive selection.[30] This is consistent with a recent emergence of these genes, as it is typical for young, novel genes to undergo adaptive evolution,[32][33][34] but it also makes it difficult to be completely sure that the candidates encode truly functional products. A subsequent study using methods similar to Levine et al. and an expressed sequence tag library derived from D. yakuba testes identified seven genes derived from six unique de novo gene birth events in D. yakuba and/or the closely related D. erecta.[29]
Three of these genes are extremely short (<90 bp), suggesting that they may be RNA genes,[29] although several examples of very short functional peptides have also been documented.[35][36][37][38] Around the same time as these studies in Drosophila were published, a homology search of genomes from all domains of life, including 18 fungal genomes, identified 132 fungal-specific proteins, 99 of which were unique to S. cerevisiae.[39]
Since these initial studies, many groups have identified specific cases of de novo gene birth events in diverse organisms.[40] The BSC4 gene in S. cerevisiae, identified in 2008, shows evidence of purifying selection, is expressed at both the mRNA and protein levels, and when deleted is synthetically lethal with two other yeast genes, all of which indicate a functional role for the BSC4 gene product.[41] Historically, one argument against the notion of widespread de novo gene birth is the evolved complexity of protein folding. Interestingly, Bsc4 was later shown to adopt a partially folded state that combines properties of native and non-native protein folding.[42] Another well-characterized example in yeast is MDF1, which both represses mating efficiency and promotes vegetative growth, and is intricately regulated by a conserved antisense ORF.[43][44] In plants, the first de novo gene to be functionally characterized was QQS, an Arabidopsis thaliana gene identified in 2009 that regulates carbon and nitrogen metabolism.[45] The first functionally characterized de novo gene identified in mice, a noncoding RNA gene, was also described in 2009.[46] In primates, a 2008 informatic analysis estimated that 15/270 primate orphan genes had been formed de novo.[47] A 2009 report identified the first three de novo human genes, one of which is a therapeutic target in chronic lymphocytic leukemia.[48] Since this time, a plethora of genome-level studies have identified large numbers of orphan genes in many organisms, although the extent to which they arose de novo, and the degree to which they can be deemed functional, remain debated.
Identification
Identification of de novo emerging sequences
There are two major approaches to the systematic identification of novel genes: genomic phylostratigraphy[49] and synteny-based methods.[50] Both approaches are widely used, individually or in a complementary fashion.
Genomic phylostratigraphy
Genomic phylostratigraphy involves examining each gene in a focal species and inferring the presence or absence of ancestral homologs through the use of the BLAST sequence alignment algorithms[51] or related tools. Each gene in the focal species can be assigned an “age” (aka “conservation level” or “genomic phylostratum”) that is based on a predetermined phylogeny, with the age corresponding to the most distantly related species in which a homolog is detected.[49] When a gene lacks any detectable homolog outside of its own genome, or close relatives, it is said to be a novel, taxonomically restricted or orphan gene, although such a designation is of course dependent on the group of species being searched against.
Phylogenetic trees are limited by the set of closely related genomes that are available, and results are dependent on BLAST search criteria.[52] Because it is based on sequence similarity, it is often difficult for phylostratigraphy to determine whether a novel gene has emerged de novo or has diverged from an ancestral gene beyond recognition, for instance following a duplication event. This was pointed out by a study that simulated the evolution of genes of equal age and found that distant orthologs can be undetectable for the most rapidly evolving genes.[53] When accounting for changes in the rate of evolution to portions of young genes that acquire selected functions, a phylostratigraphic approach was much more accurate at assigning gene ages in simulated data.[54] A subsequent pair of studies using simulated evolution found that phylostratigraphy failed to detect an ortholog in the most distantly related species for 13.9% of D. melanogaster genes and 11.4% of S. cerevisiae genes.[55][56] Similarly, a spurious relationship between a gene’s age and its likelihood to be involved in a disease process was claimed to be detected in the simulated data.[56] However, a reanalysis of studies that used phylostratigraphy in yeast, fruit flies and humans found that even when accounting for such error rates and excluding difficult-to-stratify genes from the analyses, the qualitative conclusions were unaffected for all three studies.[57] The impact of phylostratigraphic bias on studies examining various features of de novo genes (see below) remains debated.
To increase the detectability of ancestral homologues, sensitive sequence-based similarity searches, such as CS-BLAST and Hidden Markov Model (HMM)-based searches, may also be used, alone or in combination with BLAST-based phylostratigraphy analysis, to identify de novo genes. The PSI-BLAST technique[58] is particularly useful for detecting ancient homologs. A benchmarking study found that some of these “profile-based” analyses were more accurate than conventional pairwise tools.[59] The impact of false positives, when genes are incorrectly inferred to have an ancestral homolog when they are new in reality, on our understanding of de novo gene birth has not yet been specifically assessed.
It is important to disentangle the technical difficulties associated with detection of the oldest ancestor of a gene, and estimates of how old a gene is (the ultimate goal of phylostratigraphy), from challenges linked to inferring the mechanisms by which a gene has evolved.[52] Young and ancestral genes can all have evolved de novo, or through other mechanisms. The current approach of choice to determine whether a gene has emerged de novo is synteny, and can generally only be applied to young genes.[60]
Synteny-based approaches
Approaches based on the analysis of syntenic sequences in outgroups – blocks of sequence in which the order and relative positioning of features has been maintained – allow for the identification of non-genic ancestors of candidate de novo genes.[10][52] Syntenic alignments are anchored by short, conserved “markers.” Genes are the most common marker in defining syntenic blocks, although k-mers and exons are also used.[61][50] Assuming that a high-quality syntenic alignment can be obtained, confirmation that the syntenic region lacks coding potential in outgroup species allows a de novo origin to be asserted with higher confidence.[52] The strongest possible evidence for de novo emergence is the inference of the specific mutation(s) that created coding potential, typically through the analysis of microsyntenic regions of closely related species.
One challenge in applying synteny-based methods is the fact that synteny can be difficult to detect across longer timescales. To address this, various techniques have been tried, such as using exons clustered irrespective of their specific order to define syntenic blocks[50] or algorithms that use well-conserved genomic regions to expand microsyntenic blocks.[62] There are also difficulties associated with applying synteny-based approaches to genome assemblies that are fragmented[63] or in lineages with high rates of chromosomal rearrangements, as is common in insects.[64] Although synteny-based approaches have conventionally been lower-throughput in nature, they are now being applied to genome-wide surveys of de novo genes[47][48][65][66][67][68][69][70] and represent a promising area of algorithmic development for gene birth dating. Some have used synteny-based approaches in combination with similarity searches in an attempt to develop standardized, stringent pipelines[60] that can be applied to any group of genomes in an attempt to address discrepancies in the various lists of de novo genes that have been generated (see below).
Determination of status
Even when the evolutionary origin of a particular sequence has been rigorously established computationally, it is important to note that there is a lack of consensus about what constitutes a genuine de novo gene birth event. One reason for this is a lack of agreement on whether or not the entirety of the newly genic sequence must be non-genic in origin. With respect to protein-coding de novo genes, it has been proposed that de novo genes be divided into subtypes corresponding to the proportion of the ORF in question that was derived from previously noncoding sequence.[52] Furthermore, for de novo gene birth to occur, the sequence in question must not just have emerged de novo but must in fact be a gene. Accordingly, the discovery of de novo gene birth has also led to a questioning of what constitutes a gene, with some models establishing a strict dichotomy between genic and non-genic sequences, and others proposing a more fluid continuum (see below).
All definitions of genes are linked to the notion of function, as it is generally agreed that a genuine gene should encode a functional product, be it RNA or protein. There are, however, different views of what constitutes function, depending in part on whether a given sequence is assessed using genetic, biochemical, or evolutionary approaches.[52][71][72][73] The ambiguity of the concept of ‘function’ is especially problematic for the de novo gene birth field, where the objects of study are often rapidly evolving.[73] To address these challenges, the Pittsburgh Model of Function deconstructs ‘function’ into five meanings to describe the different properties that are acquired by a locus undergoing de novo gene birth : Expression, Capacities, Interactions, Physiological Implications, and Evolutionary Implications. [73]
It is generally accepted that a genuine de novo gene is expressed in at least some context,[5] allowing selection to operate, and many studies use evidence of expression as an inclusion criterion in defining de novo genes. The expression of sequences at the mRNA level may be confirmed individually through conventional techniques such as quantitative PCR, or globally through more modern techniques such as RNA sequencing (RNA-seq). Similarly, expression at the protein level can be determined with high confidence for individual proteins using techniques such as mass spectrometry or western blotting, while ribosome profiling (Ribo-seq) provides a global survey of translation in a given sample. Ideally, to confirm that the gene in question arose de novo, a lack of expression of the syntenic region of outgroup species would also be demonstrated.[74]
Confirmation of gene expression is only one approach to infer function. Genetic approaches, where one seeks to detect a specific phenotype or change in fitness upon disruption of a particular sequence, are considered by some to be the gold standard;[72] however, for large-scale analyses of entire genomes, obtaining such evidence is often not feasible. Other experimental approaches, including screens for protein-protein and/or genetic interactions, may also be employed to confirm a biological effect for a particular de novo ORF. As more is learned about a particular locus, standard molecular biology techniques can be applied to dissect its specific cellular role.
Alternatively, evolutionary approaches may be employed to infer the existence of a molecular function from computationally derived signatures of selection. In the case of TRGs, one common signature of selection is the ratio of nonsynonymous to synonymous substitutions (dN/dS ratio), calculated from different species from the same taxon. The neutral expectation for this ratio is 1; most protein-coding genes have a ratio below 1, indicating selective constraint, although a gene under strong directional selection may have a ratio above 1. A ratio below 1 is thus taken as evidence for selection against loss of function.[71] Similarly, in the case of species-specific genes, polymorphism data may be used to calculate a pN/pS ratio from different strains or populations of the focal species. Given that young, species-specific de novo genes lack deep conservation by definition, detecting statistically significant deviations from 1 can be difficult without an unrealistically large number of sequenced strains/populations. An example of this can be seen in Mus musculus, where three very young de novo genes lack signatures of selection despite well-demonstrated physiological roles.[75] For this reason, pN/pS approaches are often applied to groups of candidate genes, allowing researchers to infer that at least some of them are evolutionarily conserved, without being able to specify which. Other signatures of selection, such as the degree of nucleotide divergence within syntenic regions, conservation of ORF boundaries, or for protein-coding genes, a coding score based on nucleotide hexamer frequencies, have instead been employed.[76]
Despite these and other challenges in the identification of de novo gene birth events, there is now abundant evidence indicating that the phenomenon is not only possible, but has occurred in every lineage systematically examined thus far.[40]
Prevalence
Estimates of numbers
Estimates regarding the frequency of de novo gene birth and the number of de novo genes in various lineages vary widely and are highly dependent on methodology. Studies may identify de novo genes by phylostratigraphy/BLAST-based methods alone, or may employ a combination of computational techniques (see above), and may or may not assess experimental evidence for expression and/or biological role.[10] Furthermore, genome-scale analyses may consider all or most ORFs in the genome,[77] or may instead limit their analysis to previously annotated genes.
The D. melanogaster lineage is illustrative of these differing approaches. An early survey using a combination of BLAST searches performed on cDNA sequences along with manual searches and synteny information identified 72 new genes specific to D. melanogaster and 59 new genes specific to three of the four species in the D. melanogaster species complex. This report found that only 2/72 (~2.8%) of D. melanogaster-specific new genes and 7/59 (~11.9%) of new genes specific to the species complex were derived de novo,[69] with the remainder arising via duplication/retroposition. Similarly, an analysis of 195 young (<35 million years old) D. melanogaster genes identified from syntenic alignments found that only 16 had arisen de novo.[67] In contrast, an analysis focused on transcriptomic data from the testes of six D. melanogaster strains identified 106 fixed and 142 segregating de novo genes.[68] For many of these, ancestral ORFs were identified but were not expressed. Highlighting the differences between inter- and intra-species comparisons, a study in natural Saccharomyces paradoxus populations found that the number of de novo polypeptides identified more than doubled when considering intra-species diversity.[78] In primates, one early study identified 270 orphan genes (unique to humans, chimpanzees, and macaques), of which 15 were thought to have originated de novo,[47] while a later report identified 60 de novo genes in humans alone that are supported by transcriptional and proteomic evidence.[70] Studies in other lineages/organisms have also reached different conclusions with respect to the number of de novo genes present in each organism, as well as the specific sets of genes identified. A sample of these large-scale studies is described in the table below.
A reanalysis of three such studies in murines that identified between 69 and 773 candidate de novo genes argued that the various estimates included many genes that were not in fact de novo genes.[79] Many candidates were excluded on the basis of no longer being annotated in the major databases. A conservative approach was applied to the remaining genes, which excluded candidates with paralogs, distantly related homologs or conserved domains, or that lacked syntenic sequence information in non-rodents. This approach validated ~40% of candidate de novo genes, resulting in an upper estimate of only 11.6 de novo genes formed (and retained) per million years, a rate ~5-10 times slower than what was estimated for novel genes formed by duplication.[79] It is notable that even after application of this stringent pipeline, the 152 validated de novo genes that remained still represents a significant fraction of the mouse genome likely to have originated de novo. Generally speaking, however, it remains debated whether duplication and divergence or de novo gene birth represent the dominant mechanism for the emergence of new genes,[67][69][77][80][81][82] in part due to the fact that de novo genes are likely both to emerge and to be lost more frequently than other young genes (see below). In a study on the origin of orphan genes in 3 different eukaryotic lineages, authors found that on average only around 30% of orphan genes can be explained by sequence divergence.[82]
Dynamics
It is important to distinguish between the frequency of de novo gene birth and the number of de novo genes in a given lineage. If de novo gene birth is frequent, it might be expected that genomes would tend to grow in their gene content over time; however, the gene content of genomes is usually relatively stable.[10] This implies that a frequent gene death process must balance de novo gene birth, and indeed, de novo genes are distinguished by their rapid turnover relative to established genes. In support of this notion, recently emerged Drosophila genes are much more likely to be lost, primarily through pseudogenization, with the youngest orphans being lost at the highest rate;[83] this is despite the fact that some Drosophila orphan genes have been shown to rapidly become essential.[67] A similar trend of frequent loss among young gene families was observed in nematode genus Pristionchus.[84] Similarly, an analysis of five mammalian transcriptomes found that most ORFs in mice were either very old or species specific, implying frequent birth and death of de novo transcripts.[81] In wild S. paradoxus populations, de novo ORFs emerge and are lost at similar rates.[78] Nevertheless, there remains a positive correlation between the number of species-specific genes in a genome and the evolutionary distance from its most recent ancestor.[85] In addition to the birth and death of de novo genes at the level of the ORF, mutational and other processes also subject genomes to constant “transcriptional turnover”. One study in murines found that while all regions of the ancestral genome were transcribed at some point in at least one descendant, the portion of the genome under active transcription in a given strain or subspecies is subject to rapid change.[86] The transcriptional turnover of noncoding RNA genes is particularly fast as compared to that of coding genes.[87]
Features
Recently emerged de novo genes differ from established genes in a number of ways. Across a broad range of species, young and/or taxonomically restricted genes or ORFs have been reported to be shorter in length than established genes, to evolve more rapidly, and to be less expressed.[47][77][83][84][88][89][90][91][92][93][94][95] Although these trends are also expected to occur as a result of homology detection bias (see Genomic phylostratigraphy section above), a reanalysis of several studies that reduced this bias by removing genes whose ages are more challenging to determine found that the qualitative conclusions reached in these studies were unaffected.[57] In addition, the tendency for young genes to have fewer hydrophobic amino acids,[96] and to have them more clustered near one another along the primary sequence,[97] have been statistically controlled for evolutionary rate and for length, and so are not due to homology detection bias.
The expression of young genes has also been found to be more tissue- or condition-specific than that of established genes.[29][31][47][68][70][77][93][98][99][100] In particular, relatively high expression of de novo genes was observed in male reproductive tissues in Drosophila, mice, and humans (see below), and, in humans, in the cerebral cortex or the brain more generally.[70][101] In animals with adaptive immune systems, higher expression in the brain and testes may at least in part be a function of the immune-privileged nature of these tissues. An analysis in mice found specific expression of intergenic transcripts in the thymus and spleen (in addition to the brain and testes), and it has been proposed that in vertebrates de novo transcripts must first be expressed in these tissues before they can be expressed in tissues subject to surveillance by immune cells.[100] Older genes have more transcription factor regulation, indicative of their integration into larger molecular networks. Similarly, the likelihood of physical interactions, as well as the likelihood and strength of genetic interactions, is correlated with ORF age as determined by phylostratigraphy.[102]
Lineage-dependent features
Features of de novo genes can depend on the species or lineage being examined. This appears to partly be a result of the fact that genomes vary in their GC content, and young genes bear more similarity to non-genic sequences from the genome in which they arose than do established genes.[103] Features such as the percentage of transmembrane residues and the relative frequency of various predicted secondary structural features show a strong GC dependency in orphan genes, whereas in more ancient genes these features are only weakly influenced by GC content.[103]
The relationship between gene age and the amount of predicted intrinsic structural disorder (ISD) in the encoded proteins has been subject to considerable debate. It has been claimed that ISD is also a lineage-dependent feature, exemplified by the fact that in organisms with relatively high GC content, ranging from D. melanogaster to the parasite Leishmania major, young genes have high ISD,[104][105] while in a low GC genome such as budding yeast, several studies have shown that young genes have low ISD.[77][88][95][103] However, a study that excluded young genes with dubious evidence for functionality, defined in binary terms as being under selection for gene retention, found that the remaining young yeast genes have high ISD, suggesting that the yeast result may be due to contamination of the set of young genes with ORFs that do not meet this definition, and hence are more likely to have properties that reflect GC content and other non-genic features of the genome.[96] Beyond the very youngest orphans, this study found that ISD tends to decrease with increasing gene age, and that this is primarily due to amino acid composition rather than GC content per se.[96] Within shorter time scales, a focus on de novo genes that have the most validation suggests that younger genes are more disordered in Lachancea, but less disordered in Saccharomyces.[95]
Role of epigenetic modifications
An examination of de novo genes in A. thaliana found that they are both hypermethylated and generally depleted of histone modifications.[66] In agreement with either the proto-gene model or contamination with non-genes (see below), methylation levels of de novo genes were intermediate between established genes and intergenic regions. The methylation patterns of these de novo genes are stably inherited, and methylation levels were highest, and most similar to established genes, in de novo genes with verified protein-coding ability.[66] In the pathogenic fungus Magnaporthe oryzae, less conserved genes tend to have methylation patterns associated with low levels of transcription.[106] A study in yeasts also found that de novo genes are enriched at recombination hotspots, which tend to be nucleosome-free regions.[95]
In Pristionchus pacificus, orphan genes with confirmed expression display chromatin states that differ from those of similarly expressed established genes.[94] Orphan gene start sites have epigenetic signatures that are characteristic of enhancers, in contrast to conserved genes that exhibit classical promoters.[94] Many unexpressed orphan genes are decorated with repressive histone modifications, while a lack of such modifications facilitates transcription of an expressed subset of orphans, supporting the notion that open chromatin promotes the formation of novel genes.[94]
Models and mechanisms
Several theoretical models and possible mechanisms of de novo gene birth have been described. The models are generally not mutually exclusive, and it is possible that multiple mechanisms may give rise to de novo genes.[52]
ORF first vs. transcription first
For birth of a de novo protein-coding gene to occur, a non-genic sequence must both be transcribed and acquire an ORF before becoming translated. These events may in theory occur in either order, and there is evidence supporting both an “ORF first” and a “transcription first” model.[5] An analysis of de novo genes that are segregating in D. melanogaster with respect to their expression found that sequences that are transcribed had similar coding potential to the orthologous sequences from lines lacking evidence of transcription,[68] supporting the notion that many ORFs, at least, exist prior to being expressed. The antifreeze glycoprotein gene AFGP, which emerged de novo in Arctic codfishes, provides a more definitive example in which the de novo emergence of the ORF was shown to precede that of the promoter region.[107] Furthermore, putatively non-genic ORFs long enough to encode functional peptides are numerous in eukaryotic genomes, and expected to occur at high frequency by chance.[68][77] At the same time, transcription of eukaryotic genomes is far more extensive than previously thought, and documented examples also exist of genomic regions that were transcribed prior to the appearance of an ORF that became a de novo gene.[108] The proportion of de novo genes that are protein-coding is unknown, but the appearance of “transcription first” has led some to posit that protein-coding de novo genes may first exist as RNA gene intermediates. The case of bifunctional RNAs, which are both translated and function as RNA genes, shows that such a mechanism is plausible.[109]
The two events may occur simultaneously when chromosomal rearrangement is the event that precipates gene birth.[110]
“Out of Testis” hypothesis
An early case study of de novo gene birth, which identified five de novo genes in D. melanogaster, noted preferential expression of these genes in the testes,[30] and several additional de novo genes were identified using transcriptomic data derived from the testes and male accessory glands of D. yakuba and D. erecta[29][31] (see above). This was in keeping with the rapid evolution of genes related to reproduction that has been observed across a range of lineages,[111][112][113] suggesting that sexual selection may play a key role in adaptive evolution and de novo gene birth. A subsequent large-scale analysis of six D. melanogaster strains identified 248 testis-expressed de novo genes, of which ~57% were not fixed.[68] It has been suggested that the large number of de novo genes with male-specific expression identified in Drosophila is likely due to the fact that such genes are preferentially retained relative to other de novo genes, for reasons that are not entirely clear.[83] Interestingly, two putative de novo genes in Drosophila (Goddard and Saturn) were shown to be required for normal male fertility.[114]
In humans, a study that identified 60 human-specific de novo genes found that their average expression, as measured by RNA-seq, was highest in the testes.[70] Another study looking at mammalian-specific genes more generally also found enriched expression in the testes.[115] Transcription in mammalian testes is thought to be particularly promiscuous, due in part to elevated expression of the transcription machinery[116][117] and an open chromatin environment.[118] Along with the immune-privileged nature of the testes (see above), this promiscuous transcription is thought to create the ideal conditions for the expression of non-genic sequences required for de novo gene birth. Testes-specific expression seems to be a general feature of all novel genes, as an analysis of Drosophila and vertebrate species found that young genes showed testes-biased expression regardless of their mechanism of origination.[98]
Pervasive expression
With the development and wide use of technologies such as RNA-seq and Ribo-seq, eukaryotic genomes are now known to be pervasively transcribed[119][120][121][122] and translated.[123] Many ORFs that are either unannotated, or annotated as long non-coding RNAs (lncRNAs), are translated at some level, under at least some condition, or in a particular tissue.[77][123][124][125][126][127] Though infrequent, these translation events expose non-genic sequence to selection. This pervasive expression forms the basis for several models describing de novo gene birth.
Most non-genic ORFs that are translated appear to be evolving neutrally.[78][77][124] The preadaptation and proto-gene models both predict, however, that expression of non-genic ORFs will occasionally provide an adaptive advantage to the cell. Differential translation of proto-genes in stress conditions, as well as an enrichment near proto-genes of binding sites for transcription factors involved in regulating stress response,[77] support the adaptive potential of proto-genes. Furthermore, it is known that novel, functional proteins can be experimentally evolved from random amino acid sequences.[128] Random sequences are generally well tolerated in vivo; many readily form secondary structures, and even highly disordered proteins may take on important biological roles.[129][130][131] The pervasive nature of translation suggests that new proto-genes emerge frequently, usually returning to the non-genic state. In wild S. paradoxus populations, some ORFs with exaggerated gene-like features are found among the pool of translated intergenic polypeptides.[78] It is not clear whether such ORFs are preferentially retained.
It has been speculated that the epigenetic landscape of de novo genes in the early stages of formation may be particularly variable between and among populations, resulting in variable levels of gene expression and thereby allowing young genes to explore the “expression landscape.”[132] The QQS gene in A. thaliana is one example of this phenomenon; its expression is negatively regulated by DNA methylation that, while heritable for several generations, varies widely in its levels both among natural accessions and within wild populations.[132] Epigenetics are also largely responsible for the permissive transcriptional environment in the testes, particularly through the incorporation into nucleosomes of non-canonical histone variants that are replaced by histone-like protamines during spermatogenesis.[133]
Preadaptation model
The preadaptation model of de novo gene birth uses mathematical modeling to show that when sequences that are normally hidden are exposed to weak or shielded selection, the resulting pool of “cryptic” sequences (i.e. proto-genes) can be purged of “self-evidently deleterious” variants, such as those prone to lead to protein aggregation, and thus enriched in potential adaptations relative to a completely non-expressed and unpurged set of sequences.[134] This revealing and purging of cryptic deleterious non-genic sequences is a byproduct of pervasive transcription and translation of intergenic sequences, and is expected to facilitate the birth of functional de novo protein-coding genes.[126] This is because by eliminating the most deleterious variants, what is left is, by a process of elimination, more likely to be adaptive than expected from random sequences.
The mathematics of the preadaptation model assume that the distribution of fitness effects is bimodal, with new sequences of mutations tending to break something or tinker, but rarely in between.[134][135] From this it is derived that populations may either evolve local solutions, in which selection operates on each individual locus and a relatively high error rate is maintained, or the global solution of a low error rate which permits the accumulation of deleterious cryptic sequences.[134] De novo gene birth is thought to be favored in populations that evolve local solutions, as the relatively high error rate will result in a pool of cryptic variation that is “preadapted” through the purging of deleterious sequences. Local solutions are more likely in populations with a high effective population size.
Proto-gene model
This proto-gene model agrees with the preadaptation model about the importance of pervasive expression, and refers to the set of pervasively expressed sequences that do not meet all definitions of a gene as “proto-genes”.[77] Where it differs is that it that envisages a more gradual process under selection from non-genic to genic state, rejecting binary classification, with proto-genes expected to exhibit features intermediate between genes and non-genes.
Testable differences between models
Using the evolutionary definition of function (i.e. that a gene is by definition under purifying selection against loss), the preadaptation model assumes that “gene birth is a sudden transition to functionality”[96] that occurs as soon as an ORF acquires a net beneficial effect. In order to avoid being deleterious, newborn genes are expected to display exaggerated versions of genic features associated with the avoidance of harm. This is in contrast to the proto-gene model, which expects newborn genes to have features intermediate between old genes and non-genes.[96]
Several features of ORFs correlate with ORF age as determined by phylostratigraphic analysis (see above), with young ORFs having properties intermediate between old ORFs and non-genes; this has been taken as evidence in favor of the proto-gene model, in which proto-gene state is a continuum .[77] This evidence has been criticized, because the same apparent trends are also expected under a model in which identity as a gene is a binary. Under this model, when each age group contains a different ratio of genes vs. non-genes, Simpson's paradox can generate correlations in the wrong direction.[96]
More specifically, in support of the preadaptation model, an analysis of ISD in mice and yeast found that young genes have higher ISD than old genes, while random non-genic sequences tend to show the lowest levels of ISD.[96] Although the observed trend may have partly resulted from a subset of young genes derived by overprinting,[79] higher ISD in young genes is also seen among overlapping viral gene pairs.[136] Reaching consensus over ISD values of the very youngest genes is made difficult by different annotation standards,[81][97] as well as by disagreement over whether genes represent a binary or a continuous category.[77][96] When proto-genes with less evidence for a selected function are excluded from the data in which a continuum was seen,[77] the slope of the ISD trend is reversed.[96] However, there remains uncertainty about whether the observed trends hold consistently over shorter timescales.[81][97] With respect to other predicted structural features such as β-strand content and aggregation propensity, the peptides encoded by proto-genes are similar to non-genic sequences and categorically distinct from canonical genes.[102]
Grow slow and moult model
The “grow slow and moult” model describes a potential mechanism of de novo gene birth, particular to protein-coding genes. In this scenario, existing protein-coding ORFs expand at their ends, especially their 3’ ends, leading to the creation of novel N- and C-terminal domains.[137][138][139][140][141] Novel C-terminal domains may first evolve under weak selection via occasional expression through read-through translation, as in the preadaptation model, only later becoming constitutively expressed through a mutation that disrupts the stop codon.[134][138] Genes experiencing high translational readthrough tend to have intrinsically disordered C-termini.[142] Furthermore, existing genes are often close to repetitive sequences that encode disordered domains. These novel, disordered domains may initially confer some non-specific binding capability that becomes gradually refined by selection. Sequences encoding these novel domains may occasionally separate from their parent ORF, leading or contributing to the creation of a de novo gene.[138] Interestingly, an analysis of 32 insect genomes found that novel domains (i.e. those unique to insects) tend to evolve fairly neutrally, with only a few sites under positive selection, while their host proteins remain under purifying selection, suggesting that new functional domains emerge gradually and somewhat stochastically.[143]
Human health
In addition to its significance for the field of evolutionary biology, de novo gene birth has implications for human health. It has been speculated that novel genes, including de novo genes, may play an outsized role in species-specific traits;[6][10][40][144] however, many species-specific genes lack functional annotation.[115] Nevertheless, there is evidence to suggest that human-specific de novo genes are involved in disease processes such as cancer. NYCM, a de novo gene unique to humans and chimpanzees, regulates the pathogenesis of neuroblastomas in mouse models,[145] and the primate-specific PART1, an lncRNA gene, has been identified as both a tumor suppressor and an oncogene in different contexts.[47][146][147] Several other human- or primate-specific de novo genes, including PBOV1,[148] GR6,[149][150] MYEOV,[151] ELFN1-AS1,[152] and CLLU1,[48] are also linked to cancer. Some have even suggested considering tumor-specifically expressed, evolutionary novel genes as their own class of genetic elements, noting that many such genes are under positive selection and may be neofunctionalized in the context of tumors.[152]
The specific expression of many de novo genes in the human brain[70] also raises the intriguing possibility that de novo genes influence human cognitive traits. One such example is FLJ33706, a de novo gene that was identified in GWAS and linkage analyses for nicotine addiction and shows elevated expression in the brains of Alzheimer’s patients.[153] Generally speaking, expression of young, primate-specific genes is enriched in the fetal human brain relative to the expression of similarly young genes in the mouse brain.[154] Most of these young genes, several of which originated de novo, are expressed in the neocortex, which is thought to be responsible for many aspects of human-specific cognition. Many of these young genes show signatures of positive selection, and functional annotations indicate that they are involved in diverse molecular processes, but are enriched for transcription factors.[154]
In addition to their roles in cancer processes, de novo originated human genes have been implicated in the maintenance of pluripotency[155] and in immune function.[47][115][156] The preferential expression of de novo genes in the testes (see above) is also suggestive of a role in reproduction. Given that the function of many de novo human genes remains uncharacterized, it seems likely that an appreciation of their contribution to human health and development will continue to grow.
| Organism/Lineage | Homology Detection Method(s) | Evidence of Expression? | Evidence of Selection? | Evidence of Physiological Role? | # Orphan/De Novo Genes | Notes | Ref. |
|---|---|---|---|---|---|---|---|
| Arthropods | BLASTP for all 30 species against each other, TBLASTN for Formicidae only, searched by synteny for unannotated orthologs in Formicidae only | ESTs, RNA-seq; RT-PCR on select candidates | 37 Formicidae-restricted orthologs appear under positive selection (M1a to M2a and M7 to M8 models using likelihood ratio tests); as a group, Formicidae-restricted orthologs have a significantly higher Ka/Ks rate than non-restricted orthologs | Prediction of signal peptides and subcellular localization for subset of orphans | ~65,000 orphan genes across 30 species | Abundance of orphan genes dependent on time since emergence from common ancestor; >40% of orphans from intergenic matches indicating possible de novo origin | [85] |
| Arabidopsis thaliana | BLASTP against 62 species, PSI-BLAST against NCBI nonredundant protein database, TBLASTN against PlantGDB-assembled unique transcripts database; searched syntenic region of two closely related species | Transcriptomic and translatomic data from multiple sources | Allele frequencies of de novo genes correlated with their DNA methylation levels | None | 782 de novo genes | Also assessed DNA methylation and histone modifications | [66] |
| Bombyx mori | BLASTP against four lepidopterans, TBLASTN against lepidopteran EST sequences, BLASTP against NCBI nonredundant protein database | Microarray, RT-PCR | None | RNAi on five de novo genes produced no visible phenotypes | 738 orphan genes | Five orphans identified as de novo genes | [92] |
| Brassicaceae | BLASTP against NCBI nonredundant protein database, TBLASTN against NCBI nucleotide database, TBLASTN against NCBI EST database, PSI-BLAST against NCBI nonredundant protein database, InterProScan[157] | Microarray | None | TRGs enriched for expression changes in response to abiotic stresses compared to other genes | 1761 nuclear TRGs; 28 mitochondrial TRGs | ~2% of TRGs thought to be de novo genes | [93] |
| Drosophila melanogaster | BLASTN of query cDNAs against D. melanogaster, D. simulans and D. yakuba genomes; also performed check of syntenic region in sister species | cDNA/ expressed sequence tags (ESTs) | Ka/Ks ratios calculated between retained new genes and their parental genes are significantly >1, indicating most new genes are functionally constrained | List includes several genes with characterized molecular roles | 72 orphan genes; 2 de novo genes | Gene duplication dominant mechanism for new genes; 7/59 orphans specific to D. melanogaster species complex identified as de novo | [69] |
| Drosophila melanogaster | Presence or absence of orthologs in other Drosophila species inferred by synteny based on UCSC genome alignments and FlyBase protein-based synteny; TBLASTN against Drosophila subgroup | Indirect (RNAi) | Youngest essential genes show signatures of positive selection (α=0.25 as a group) | Knockdown with constitutive RNAi lethal for 59 TRGs | 195 “young” (>35myo) TRGs; 16 de novo genes | Gene duplication dominant mechanism for new genes | [67] |
| Drosophila melanogaster | RNA-seq in D. melanogaster and close relatives; syntenic alignments with D. simulans and D. yakuba; BLASTP against NCBI nonredundant protein database | RNA-seq | Nucleotide diversity lower in non-expressing relatives; Hudson-Kreitman-Aguade-like statistic lower in fixed de novo genes than in intergenic regions | Structural features of de novo genes (e.g. enrichment of long ORFs) suggestive of function | 106 fixed and 142 segregating de novo genes | Specifically expressed in testes | [68] |
| Homo sapiens | BLASTP against other primates; BLAT against chimpanzee and orangutan genomes, manual check of syntenic regions in chimpanzee and orangutan | RNA-seq | Substitution rate provides some evidence for weak selection; 59/60 de novo genes are fixed | None | 60 de novo genes | Enabling mutations identified; highest expression seen in brain and testes | [70] |
| Homo sapiens | BLASTP against chimpanzee, BLAT and Search of syntenic region in chimpanzee, manual check of syntenic regions in chimpanzee and macaque | EST/cDNA | No evidence of selective constraint seen by nucleotide divergence | One of the genes identified has a known role in leukemia | 3 de novo genes | Estimated that human genome contains ~ 18 human-specific de novo genes | [48] |
| Lachancea and Saccharomyces | BLASTP of all focal species against each other, BLASTP against NCBI nonredundant protein database, PSI-BLAST against NCBI nonredundant protein database, HMM Profile-Profile of TRG families against each other; families then merged and searched against four profile databases | Mass Spectrometry (MS) | Ka/Ks ratios across Saccharomyces indicate that candidates are under weak selection that increases with gene age; in Lachancea species with multiple strains, pN/pS ratios are lower for de novo candidates than for "spurious TRGs" | None | 288 candidate de novo TRGs in Saccharomyces, 415 in Lachancea | MS evidence of translation for 25 candidates | [95] |
| Mus musculus and Rattus norvegicus | BLASTP of rat and mouse against each other, BLASTP against Ensembl compara database; searched syntenic regions in rat and mouse | UniGene Database | Subset of genes shows low nucleotide diversity and high ORF conservation across 17 strains | Two mouse genes cause morbidity when knocked out | 69 de novo genes in mouse and 6 "de novo" genes in rat | Enabling mutations identified for 9 mouse genes | [158] |
| Mus musculus | BLASTP against NCBI nonredundant protein database | Microarray | None | None | 781 orphan genes | Age-dependent features of genes compatible with de novo emergence of many orphans | [80] |
| Oryza | Protein-to-protein and nucleotide-to-nucleotide BLAT against eight Oryza species and two outgroup species; searched syntenic regions of these species for coding potential | RNA-seq (all de novo TRGs); Ribosome Profiling and targeted MS (some de novo TRGs) | 22 de novo candidates appear under negative selection, and six under positive selection, as measured by Ka/Ks rate | Expression of de novo TRGs is tissue-specific | 175 de novo TRGs | ~57% of de novo genes have translational evidence; transcription predates coding potential in most cases | [159] |
| Primates | BLASTP against 15 eukaryotes, BLASTN against human genome, analysis of syntenic regions | ESTs | Ka/Ks ratios for TRGs below one but higher than established genes; coding scores consistent with translated proteins | Several genes have well-characterized cellular roles | 270 TRGs | ~5.5% of TRGs estimated to have originated de novo | [47] |
| Pristionchus pacificus | BLASTP and tBLASTN, syntenic analysis | RNA-Seq | 2 cases complete de novo gene origination | 27 other high-confidence orphans whose methods of origin included annotation artifacts, chimeric origin, alternative reading frame usage, and gene splitting with subsequent gain of de novo exons | [160] | ||
| Rodentia | BLASTP against NCBI nonredundant protein database | None | Mouse genes share 50% identity with rat ortholog | None | 84 TRGs | Species-specific genes excluded from analysis; results robust to evolutionary rate | [96] |
| Saccharomyces cerevisiae | BLASTP and PSI-BLAST against 18 fungal species, HMMER and HHpred against several databases, TBLASTN against three close relatives | None | None | Majority of orphans have characterized fitness effects | 188 orphan genes | Ages of genes determined at level of individual residues | [88] |
| Saccharomyces cerevisiae | BLASTP, TBLASTX, and TBLASTN against 14 other yeast species, BLASTP against NCBI nonredundant protein database | Ribosome Profiling | All 25 de novo genes, 115 proto-genes under purifying selection (pN/pS < 1) | None | 25 de novo genes; 1,891 “proto-genes” | De novo gene birth more common than new genes from duplication; proto-genes are unique to Saccharomyces (Sensu stricto) yeasts | [77] |
| Saccharomyces cerevisiae | BLASTN, TBLASTX, against nt/nr, manual inspection of syntenic alignment | transcripts believed to be non-coding, manual inspection of ribosome profiling traces | None | None | 1 de novo candidate gene, 217 ribosome-associated transcripts | Candidate de novo gene is polymorphic. Ribosomal profiling data is the same as in [77] | [126] |
| Saccharomyces sensu strictu | BLASTP against NCBI nonredundant protein database, TBLASTN against ten outgroup species; BLASTP and phmmer against 20 yeast species reannotated using syntenic alignments | Transcript isoform sequencing (TIF-seq), Ribosome Profiling | Most genes weakly constrained but a subset under strong selection, according to Neutrality Index, Direction of Selection, Ka/Ks, and McDonald-Kreitman tests | Subcellular localization demonstrated for five genes | ~13,000 de novo genes | >65% of de novo genes are isoforms of ancient genes; >97% from TIF-seq dataset | [65] |
Note: For purposes of this table, genes are defined as orphan genes (when species-specific) or TRGs (when limited to a closely related group of species) when the mechanism of origination has not been investigated, and as de novo genes when de novo origination has been inferred, irrespective of method of inference. The designation of de novo genes as “candidates” or “proto-genes” reflects the language used by the authors of the respective studies.
References
![]() This article was adapted from the following source under a CC BY 4.0 license (2019) (reviewer reports): "De novo gene birth". PLOS Genetics. 15 (5): e1008160. 23 May 2019. doi:10.1371/JOURNAL.PGEN.1008160. ISSN 1553-7390. PMC 6542195. PMID 31120894. Wikidata Q86320144.
This article was adapted from the following source under a CC BY 4.0 license (2019) (reviewer reports): "De novo gene birth". PLOS Genetics. 15 (5): e1008160. 23 May 2019. doi:10.1371/JOURNAL.PGEN.1008160. ISSN 1553-7390. PMC 6542195. PMID 31120894. Wikidata Q86320144.
- Long M, Betrán E, Thornton K, Wang W (November 2003). "The origin of new genes: glimpses from the young and old". Nature Reviews Genetics. 4 (11): 865–75. doi:10.1038/nrg1204. PMID 14634634. S2CID 33999892.
- Wang W, Yu H, Long M (May 2004). "Duplication-degeneration as a mechanism of gene fission and the origin of new genes in Drosophila species". Nature Genetics. 36 (5): 523–7. doi:10.1038/ng1338. PMID 15064762.
- Levy, Adam (16 October 2019). "How evolution builds genes from scratch - Scientists long assumed that new genes appear when evolution tinkers with old ones. It turns out that natural selection is much more creative". Nature. 574 (7778): 314–316. doi:10.1038/d41586-019-03061-x. PMID 31619796.
- Schmitz JF, Bornberg-Bauer E (2017). "de novo from previously non-coding DNA". F1000Research. 6: 57. doi:10.12688/f1000research.10079.1. PMC 5247788. PMID 28163910.
- Schlötterer C (April 2015). "Genes from scratch--the evolutionary fate of de novo genes". Trends in Genetics. 31 (4): 215–9. doi:10.1016/j.tig.2015.02.007. PMC 4383367. PMID 25773713.
- Kaessmann H (October 2010). "Origins, evolution, and phenotypic impact of new genes". Genome Research. 20 (10): 1313–26. doi:10.1101/gr.101386.109. PMC 2945180. PMID 20651121.
- Jacob F (June 1977). "Evolution and tinkering". Science. 196 (4295): 1161–6. Bibcode:1977Sci...196.1161J. doi:10.1126/science.860134. PMID 860134. S2CID 29756896.
- Carvunis, Anne-Ruxandra; Oss, Stephen Branden Van (2019-05-23). "De novo gene birth". PLOS Genetics. 15 (5): e1008160. doi:10.1371/journal.pgen.1008160. ISSN 1553-7404. PMC 6542195. PMID 31120894.
- Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TC (September 2009). "More than just orphans: are taxonomically-restricted genes important in evolution?". Trends in Genetics. 25 (9): 404–13. doi:10.1016/j.tig.2009.07.006. PMID 19716618.
- Tautz D, Domazet-Lošo T (August 2011). "The evolutionary origin of orphan genes". Nature Reviews Genetics. 12 (10): 692–702. doi:10.1038/nrg3053. PMID 21878963. S2CID 31738556.
- Ohno S (1970) Evolution by Gene Duplication Allen & Unwin; Springer-Verlag
- Tautz D (2014). "The discovery of de novo gene evolution". Perspectives in Biology and Medicine. 57 (1): 149–61. doi:10.1353/pbm.2014.0006. hdl:11858/00-001M-0000-0024-3416-1. PMID 25345708. S2CID 29552265.
- Grassé P-P (1977) Evolution of living organisms : evidence for a new theory of transformation Academic Press
- Barrell BG, Air GM, Hutchison CA (November 1976). "Overlapping genes in bacteriophage phiX174". Nature. 264 (5581): 34–41. Bibcode:1976Natur.264...34B. doi:10.1038/264034a0. PMID 1004533. S2CID 4264796.
- Shaw DC, Walker JE, Northrop FD, Barrell BG, Godson GN, Fiddes JC (April 1978). "Gene K, a new overlapping gene in bacteriophage G4". Nature. 272 (5653): 510–5. Bibcode:1978Natur.272..510S. doi:10.1038/272510a0. PMID 692656. S2CID 4218777.
- Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes CA, et al. (February 1977). "Nucleotide sequence of bacteriophage phi X174 DNA". Nature. 265 (5596): 687–95. Bibcode:1977Natur.265..687S. doi:10.1038/265687a0. PMID 870828. S2CID 4206886.
- Keese PK, Gibbs A (October 1992). "Origins of genes: "big bang" or continuous creation?". Proceedings of the National Academy of Sciences of the United States of America. 89 (20): 9489–93. Bibcode:1992PNAS...89.9489K. doi:10.1073/pnas.89.20.9489. PMC 50157. PMID 1329098.
- Ohno S (April 1984). "Birth of a unique enzyme from an alternative reading frame of the preexisted, internally repetitious coding sequence". Proceedings of the National Academy of Sciences of the United States of America. 81 (8): 2421–5. Bibcode:1984PNAS...81.2421O. doi:10.1073/pnas.81.8.2421. PMC 345072. PMID 6585807.
- Sabath N, Wagner A, Karlin D (December 2012). "Evolution of viral proteins originated de novo by overprinting". Molecular Biology and Evolution. 29 (12): 3767–80. doi:10.1093/molbev/mss179. PMC 3494269. PMID 22821011.
- Makałowska I, Lin CF, Hernandez K (October 2007). "Birth and death of gene overlaps in vertebrates". BMC Evolutionary Biology. 7: 193. doi:10.1186/1471-2148-7-193. PMC 2151771. PMID 17939861.
- Samandi S, Roy AV, Delcourt V, Lucier JF, Gagnon J, Beaudoin MC, et al. (October 2017). "Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins". eLife. 6. doi:10.7554/eLife.27860. PMC 5703645. PMID 29083303.
- Khan, YA; Jungreis, I; Wright, JC; Mudge, JM; Choudhary, JS; Firth, AE; Kellis, M (6 March 2020). "Evidence for a novel overlapping coding sequence in POLG initiated at a CUG start codon". BMC Genetics. 21 (1): 25. doi:10.1186/s12863-020-0828-7. PMC 7059407. PMID 32138667.
- Makałowski W, Mitchell GA, Labuda D (June 1994). "Alu sequences in the coding regions of mRNA: a source of protein variability". Trends in Genetics. 10 (6): 188–93. doi:10.1016/0168-9525(94)90254-2. PMID 8073532.
- Sorek R (October 2007). "The birth of new exons: mechanisms and evolutionary consequences". RNA. 13 (10): 1603–8. doi:10.1261/rna.682507. PMC 1986822. PMID 17709368.
- Dorit RL, Gilbert W (December 1991). "The limited universe of exons". Current Opinion in Genetics & Development. 1 (4): 464–9. doi:10.1016/S0959-437X(05)80193-5. PMID 1822278.
- Chothia C (June 1992). "Proteins. One thousand families for the molecular biologist". Nature. 357 (6379): 543–4. Bibcode:1992Natur.357..543C. doi:10.1038/357543a0. PMID 1608464. S2CID 4355476.
- Oliver SG, van der Aart QJ, Agostoni-Carbone ML, Aigle M, Alberghina L, Alexandraki D, et al. (May 1992). "The complete DNA sequence of yeast chromosome III". Nature. 357 (6373): 38–46. Bibcode:1992Natur.357...38O. doi:10.1038/357038a0. PMID 1574125. S2CID 4271784.
- Dujon B (July 1996). "The yeast genome project: what did we learn?". Trends in Genetics. 12 (7): 263–70. doi:10.1016/0168-9525(96)10027-5. PMID 8763498.
- Begun DJ, Lindfors HA, Kern AD, Jones CD (June 2007). "Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade". Genetics. 176 (2): 1131–7. doi:10.1534/genetics.106.069245. PMC 1894579. PMID 17435230.
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ (June 2006). "Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression". Proceedings of the National Academy of Sciences of the United States of America. 103 (26): 9935–9. Bibcode:2006PNAS..103.9935L. doi:10.1073/pnas.0509809103. PMC 1502557. PMID 16777968.
- Begun DJ, Lindfors HA, Thompson ME, Holloway AK (March 2006). "Recently evolved genes identified from Drosophila yakuba and D. erecta accessory gland expressed sequence tags". Genetics. 172 (3): 1675–81. doi:10.1534/genetics.105.050336. PMC 1456303. PMID 16361246.
- Betrán E, Long M (July 2003). "Dntf-2r, a young Drosophila retroposed gene with specific male expression under positive Darwinian selection". Genetics. 164 (3): 977–88. PMC 1462638. PMID 12871908.
- Jones CD, Begun DJ (August 2005). "Parallel evolution of chimeric fusion genes". Proceedings of the National Academy of Sciences of the United States of America. 102 (32): 11373–8. Bibcode:2005PNAS..10211373J. doi:10.1073/pnas.0503528102. PMC 1183565. PMID 16076957.
- Long M, Langley CH (April 1993). "Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila". Science. 260 (5104): 91–5. Bibcode:1993Sci...260...91L. doi:10.1126/science.7682012. PMID 7682012.
- Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP (May 2007). "Peptides encoded by short ORFs control development and define a new eukaryotic gene family". PLOS Biology. 5 (5): e106. doi:10.1371/journal.pbio.0050106. PMC 1852585. PMID 17439302.
- Hsu PY, Benfey PN (May 2018). "Small but Mighty: Functional Peptides Encoded by Small ORFs in Plants". Proteomics. 18 (10): e1700038. doi:10.1002/pmic.201700038. PMID 28759167.
- Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel-Duby R, Olson EN (January 2016). "A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle". Science. 351 (6270): 271–5. Bibcode:2016Sci...351..271N. doi:10.1126/science.aad4076. PMC 4892890. PMID 26816378.
- Andrews SJ, Rothnagel JA (March 2014). "Emerging evidence for functional peptides encoded by short open reading frames". Nature Reviews Genetics. 15 (3): 193–204. doi:10.1038/nrg3520. PMID 24514441.
- Nishida H (November 2006). "Detection and characterization of fungal-specific proteins in Saccharomyces cerevisiae". Bioscience, Biotechnology, and Biochemistry. 70 (11): 2646–52. doi:10.1271/bbb.60251. PMID 17090923. S2CID 11035512.
- McLysaght A, Guerzoni D (September 2015). "New genes from non-coding sequence: the role of de novo protein-coding genes in eukaryotic evolutionary innovation". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370 (1678): 20140332. doi:10.1098/rstb.2014.0332. PMC 4571571. PMID 26323763.
- Cai J, Zhao R, Jiang H, Wang W (May 2008). "De novo origination of a new protein-coding gene in Saccharomyces cerevisiae". Genetics. 179 (1): 487–96. doi:10.1534/genetics.107.084491. PMC 2390625. PMID 18493065.
- Bungard D, Copple JS, Yan J, Chhun JJ, Kumirov VK, Foy SG, et al. (November 2017). "Foldability of a Natural De Novo Evolved Protein". Structure. 25 (11): 1687–1696.e4. doi:10.1016/j.str.2017.09.006. PMC 5677532. PMID 29033289.
- Li D, Dong Y, Jiang Y, Jiang H, Cai J, Wang W (April 2010). "A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand". Cell Research. 20 (4): 408–20. doi:10.1038/cr.2010.31. PMID 20195295.
- Li D, Yan Z, Lu L, Jiang H, Wang W (December 2014). "Pleiotropy of the de novo-originated gene MDF1". Scientific Reports. 4: 7280. Bibcode:2014NatSR...4E7280L. doi:10.1038/srep07280. PMC 4250933. PMID 25452167.
- Li L, Foster CM, Gan Q, Nettleton D, James MG, Myers AM, et al. (May 2009). "Identification of the novel protein QQS as a component of the starch metabolic network in Arabidopsis leaves". The Plant Journal. 58 (3): 485–98. doi:10.1111/j.1365-313X.2009.03793.x. PMID 19154206.
- Heinen TJ, Staubach F, Häming D, Tautz D (September 2009). "Emergence of a new gene from an intergenic region". Current Biology. 19 (18): 1527–31. doi:10.1016/j.cub.2009.07.049. PMID 19733073. S2CID 12446879.
- Toll-Riera M, Bosch N, Bellora N, Castelo R, Armengol L, Estivill X, et al. (March 2009). "Origin of primate orphan genes: a comparative genomics approach". Molecular Biology and Evolution. 26 (3): 603–12. doi:10.1093/molbev/msn281. PMID 19064677.
- Knowles DG, McLysaght A (October 2009). "Recent de novo origin of human protein-coding genes". Genome Research. 19 (10): 1752–9. doi:10.1101/gr.095026.109. PMC 2765279. PMID 19726446.
- Domazet-Loso T, Brajković J, Tautz D (November 2007). "A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages". Trends in Genetics. 23 (11): 533–9. doi:10.1016/j.tig.2007.08.014. PMID 18029048.
- Gehrmann T, Reinders MJ (November 2015). "Proteny: discovering and visualizing statistically significant syntenic clusters at the proteome level". Bioinformatics. 31 (21): 3437–44. doi:10.1093/bioinformatics/btv389. PMC 4612220. PMID 26116928.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (October 1990). "Basic local alignment search tool". Journal of Molecular Biology. 215 (3): 403–10. doi:10.1016/S0022-2836(05)80360-2. PMID 2231712.
- McLysaght A, Hurst LD (September 2016). "Open questions in the study of de novo genes: what, how and why". Nature Reviews Genetics. 17 (9): 567–78. doi:10.1038/nrg.2016.78. PMID 27452112. S2CID 6033249.
- Elhaik E, Sabath N, Graur D (January 2006). "The "inverse relationship between evolutionary rate and age of mammalian genes" is an artifact of increased genetic distance with rate of evolution and time of divergence". Molecular Biology and Evolution. 23 (1): 1–3. doi:10.1093/molbev/msj006. PMID 16151190.
- Albà MM, Castresana J (April 2007). "On homology searches by protein Blast and the characterization of the age of genes". BMC Evolutionary Biology. 7: 53. doi:10.1186/1471-2148-7-53. PMC 1855329. PMID 17408474.
- Moyers BA, Zhang J (May 2016). "Evaluating Phylostratigraphic Evidence for Widespread De Novo Gene Birth in Genome Evolution". Molecular Biology and Evolution. 33 (5): 1245–56. doi:10.1093/molbev/msw008. PMC 5010002. PMID 26758516.
- Moyers BA, Zhang J (January 2015). "Phylostratigraphic bias creates spurious patterns of genome evolution". Molecular Biology and Evolution. 32 (1): 258–67. doi:10.1093/molbev/msu286. PMC 4271527. PMID 25312911.
- Domazet-Lošo T, Carvunis AR, Albà MM, Šestak MS, Bakaric R, Neme R, et al. (April 2017). "No Evidence for Phylostratigraphic Bias Impacting Inferences on Patterns of Gene Emergence and Evolution". Molecular Biology and Evolution. 34 (4): 843–856. doi:10.1093/molbev/msw284. PMC 5400388. PMID 28087778.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (September 1997). "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs". Nucleic Acids Research. 25 (17): 3389–402. doi:10.1093/nar/25.17.3389. PMC 146917. PMID 9254694.
- Saripella GV, Sonnhammer EL, Forslund K (September 2016). "Benchmarking the next generation of homology inference tools". Bioinformatics. 32 (17): 2636–41. doi:10.1093/bioinformatics/btw305. PMC 5013910. PMID 27256311.
- Vakirlis N, McLysaght A (2019). "Computational Prediction of De Novo Emerged Protein-Coding Genes". Computational Methods in Protein Evolution. Methods in Molecular Biology. 1851. pp. 63–81. doi:10.1007/978-1-4939-8736-8_4. ISBN 978-1-4939-8735-1. PMID 30298392.
- Ghiurcuta CG, Moret BM (June 2014). "Evaluating synteny for improved comparative studies". Bioinformatics. 30 (12): i9-18. doi:10.1093/bioinformatics/btu259. PMC 4058928. PMID 24932010.
- Jean G, Nikolski M (2011). "SyDiG: uncovering Synteny in Distant Genomes" (PDF). International Journal of Bioinformatics Research and Applications. 7 (1): 43–62. doi:10.1504/IJBRA.2011.039169. PMID 21441096.
- Liu D, Hunt M, Tsai IJ (January 2018). "Inferring synteny between genome assemblies: a systematic evaluation". BMC Bioinformatics. 19 (1): 26. doi:10.1186/s12859-018-2026-4. PMC 5791376. PMID 29382321.
- Ranz JM, Casals F, Ruiz A (February 2001). "How malleable is the eukaryotic genome? Extreme rate of chromosomal rearrangement in the genus Drosophila". Genome Research. 11 (2): 230–9. doi:10.1101/gr.162901. PMC 311025. PMID 11157786.
- Lu TC, Leu JY, Lin WC (November 2017). "A Comprehensive Analysis of Transcript-Supported De Novo Genes in Saccharomyces sensu stricto Yeasts". Molecular Biology and Evolution. 34 (11): 2823–2838. doi:10.1093/molbev/msx210. PMC 5850716. PMID 28981695.
- Li ZW, Chen X, Wu Q, Hagmann J, Han TS, Zou YP, Ge S, Guo YL (August 2016). "On the Origin of De Novo Genes in Arabidopsis thaliana Populations". Genome Biology and Evolution. 8 (7): 2190–202. doi:10.1093/gbe/evw164. PMC 4987118. PMID 27401176.
- Chen S, Zhang YE, Long M (December 2010). "New genes in Drosophila quickly become essential". Science. 330 (6011): 1682–5. Bibcode:2010Sci...330.1682C. doi:10.1126/science.1196380. PMC 7211344. PMID 21164016. S2CID 7899890.
- Zhao L, Saelao P, Jones CD, Begun DJ (February 2014). "Origin and spread of de novo genes in Drosophila melanogaster populations". Science. 343 (6172): 769–72. Bibcode:2014Sci...343..769Z. doi:10.1126/science.1248286. PMC 4391638. PMID 24457212.
- Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, Zhan Z, et al. (September 2008). "On the origin of new genes in Drosophila". Genome Research. 18 (9): 1446–55. doi:10.1101/gr.076588.108. PMC 2527705. PMID 18550802.
- Wu DD, Irwin DM, Zhang YP (November 2011). "De novo origin of human protein-coding genes". PLOS Genetics. 7 (11): e1002379. doi:10.1371/journal.pgen.1002379. PMC 3213175. PMID 22102831.
- Doolittle WF, Brunet TD, Linquist S, Gregory TR (May 2014). "Distinguishing between "function" and "effect" in genome biology". Genome Biology and Evolution. 6 (5): 1234–7. doi:10.1093/gbe/evu098. PMC 4041003. PMID 24814287.
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, et al. (April 2014). "Defining functional DNA elements in the human genome". Proceedings of the National Academy of Sciences of the United States of America. 111 (17): 6131–8. Bibcode:2014PNAS..111.6131K. doi:10.1073/pnas.1318948111. PMC 4035993. PMID 24753594.
- Keeling, DM; Garza, P; Nartey, CM; Carvunis, AR (1 November 2019). "The meanings of 'function' in biology and the problematic case of de novo gene emergence". eLife. 8. doi:10.7554/eLife.47014. PMC 6824840. PMID 31674305.
- Andersson DI, Jerlström-Hultqvist J, Näsvall J (June 2015). "Evolution of new functions de novo and from preexisting genes". Cold Spring Harbor Perspectives in Biology. 7 (6): a017996. doi:10.1101/cshperspect.a017996. PMC 4448608. PMID 26032716.
- Xie C, Bekpen C, Künzel S, Keshavarz M, Krebs-Wheaton R, Skrabar N, et al. (January 2019). "Studying the dawn of de novo gene emergence in mice reveals fast integration of new genes into functional networks". bioRxiv. bioRxiv 10.1101/510214. doi:10.1101/510214.
- Ruiz-Orera J, Hernandez-Rodriguez J, Chiva C, Sabidó E, Kondova I, Bontrop R, et al. (December 2015). "Origins of De Novo Genes in Human and Chimpanzee". PLOS Genetics. 11 (12): e1005721. arXiv:1507.07744. Bibcode:2015arXiv150707744R. doi:10.1371/journal.pgen.1005721. PMC 4697840. PMID 26720152.
- Carvunis AR, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, et al. (July 2012). "Proto-genes and de novo gene birth". Nature. 487 (7407): 370–4. Bibcode:2012Natur.487..370C. doi:10.1038/nature11184. PMC 3401362. PMID 22722833.
- Durand, É; Gagnon-Arsenault, I; Hallin, J; Hatin, I; Dubé, AK; Nielly-Thibault, L; Namy, O; Landry, CR (June 2019). "Turnover of ribosome-associated transcripts from de novo ORFs produces gene-like characteristics available for de novo gene emergence in wild yeast populations". Genome Research. 29 (6): 932–943. doi:10.1101/gr.239822.118. PMC 6581059. PMID 31152050.
- Casola C (2018). "From de novo to "de nono": most novel protein coding genes identified with phylostratigraphy represent old genes or recent duplicates". bioRxiv. bioRxiv 10.1101/287193. doi:10.1101/287193.
- Neme R, Tautz D (February 2013). "Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution". BMC Genomics. 14: 117. doi:10.1186/1471-2164-14-117. PMC 3616865. PMID 23433480.
- Schmitz JF, Ullrich KK, Bornberg-Bauer E (October 2018). "Incipient de novo genes can evolve from frozen accidents that escaped rapid transcript turnover". Nature Ecology & Evolution. 2 (10): 1626–1632. doi:10.1038/s41559-018-0639-7. PMID 30201962. S2CID 52181376.
- Vakirlis, N; Carvunis, AR; McLysaght, A (18 February 2020). "Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes". eLife. 9. doi:10.7554/eLife.53500. PMC 7028367. PMID 32066524.
- Palmieri N, Kosiol C, Schlötterer C (February 2014). "The life cycle of Drosophila orphan genes". eLife. 3: e01311. arXiv:1401.4956. Bibcode:2014arXiv1401.4956P. doi:10.7554/eLife.01311. PMC 3927632. PMID 24554240.
- Prabh N, Roeseler W, Witte H, Eberhardt G, Sommer RJ, Rödelsperger C (November 2018). "Pristionchus nematodes". Genome Research. 28 (11): 1664–1674. doi:10.1101/gr.234971.118. PMC 6211646. PMID 30232197.
- Wissler L, Gadau J, Simola DF, Helmkampf M, Bornberg-Bauer E (2013). "Mechanisms and dynamics of orphan gene emergence in insect genomes". Genome Biology and Evolution. 5 (2): 439–55. doi:10.1093/gbe/evt009. PMC 3590893. PMID 23348040.
- Neme R, Tautz D (February 2016). "Fast turnover of genome transcription across evolutionary time exposes entire non-coding DNA to de novo gene emergence". eLife. 5: e09977. doi:10.7554/eLife.09977. PMC 4829534. PMID 26836309.
- Kutter C, Watt S, Stefflova K, Wilson MD, Goncalves A, Ponting CP, Odom DT, Marques AC (2012). "Rapid turnover of long noncoding RNAs and the evolution of gene expression". PLOS Genetics. 8 (7): e1002841. doi:10.1371/journal.pgen.1002841. PMC 3406015. PMID 22844254.
- Ekman D, Elofsson A (February 2010). "Identifying and quantifying orphan protein sequences in fungi". Journal of Molecular Biology. 396 (2): 396–405. doi:10.1016/j.jmb.2009.11.053. PMID 19944701.
- Domazet-Loso T, Tautz D (October 2003). "An evolutionary analysis of orphan genes in Drosophila". Genome Research. 13 (10): 2213–9. doi:10.1101/gr.1311003. PMC 403679. PMID 14525923.
- Guo WJ, Li P, Ling J, Ye SP (2007). "Significant comparative characteristics between orphan and nonorphan genes in the rice (Oryza sativa L.) genome". Comparative and Functional Genomics. 2007: 21676. doi:10.1155/2007/21676. PMC 2216055. PMID 18273382.
- Wolf YI, Novichkov PS, Karev GP, Koonin EV, Lipman DJ (May 2009). "The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages". Proceedings of the National Academy of Sciences of the United States of America. 106 (18): 7273–80. doi:10.1073/pnas.0901808106. PMC 2666616. PMID 19351897.
- Sun W, Zhao XW, Zhang Z (September 2015). "Identification and evolution of the orphan genes in the domestic silkworm, Bombyx mori". FEBS Letters. 589 (19 Pt B): 2731–8. doi:10.1016/j.febslet.2015.08.008. PMID 26296317.
- Donoghue MT, Keshavaiah C, Swamidatta SH, Spillane C (February 2011). "Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana". BMC Evolutionary Biology. 11: 47. doi:10.1186/1471-2148-11-47. PMC 3049755. PMID 21332978.
- Werner MS, Sieriebriennikov B, Prabh N, Loschko T, Lanz C, Sommer RJ (November 2018). "Young genes have distinct gene structure, epigenetic profiles, and transcriptional regulation". Genome Research. 28 (11): 1675–1687. doi:10.1101/gr.234872.118. PMC 6211652. PMID 30232198.
- Vakirlis N, Hebert AS, Opulente DA, Achaz G, Hittinger CT, Fischer G, Coon JJ, Lafontaine I (March 2018). "A Molecular Portrait of De Novo Genes in Yeasts". Molecular Biology and Evolution. 35 (3): 631–645. doi:10.1093/molbev/msx315. PMC 5850487. PMID 29220506.
- Wilson BA, Foy SG, Neme R, Masel J (June 2017). "Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth". Nature Ecology & Evolution. 1 (6): 0146–146. doi:10.1038/s41559-017-0146. PMC 5476217. PMID 28642936.
- Foy SG, Wilson BA, Bertram J, Cordes MH, Masel J (April 2019). "A Shift in Aggregation Avoidance Strategy Marks a Long-Term Direction to Protein Evolution". Genetics. 211 (4): 1345–1355. doi:10.1534/genetics.118.301719. PMC 6456324. PMID 30692195.
- Zhang, JY; Zhou, Q (1 January 2019). "On the Regulatory Evolution of New Genes Throughout Their Life History". Molecular Biology and Evolution. 36 (1): 15–27. doi:10.1093/molbev/msy206. PMID 30395322. S2CID 53216993.
- Wu B, Knudson A (July 2018). "De Novo Origin of Protein-Coding Genes in Yeast". mBio. 9 (4). doi:10.1128/mBio.01024-18. PMC 6069113. PMID 30065088.
- Bekpen C, Xie C, Tautz D (August 2018). "Dealing with the adaptive immune system during de novo evolution of genes from intergenic sequences". BMC Evolutionary Biology. 18 (1): 121. doi:10.1186/s12862-018-1232-z. PMC 6091031. PMID 30075701.
- Pertea M, Shumate A, Pertea G, Varabyou A, Chang YC, Madugundu A, et al. (2018). "Thousands of large-scale RNA sequencing experiments yield a comprehensive new human gene list and reveal extensive transcriptional noise". bioRxiv. bioRxiv 10.1101/332825. doi:10.1101/332825.
- Abrusán G (December 2013). "Integration of new genes into cellular networks, and their structural maturation". Genetics. 195 (4): 1407–17. doi:10.1534/genetics.113.152256. PMC 3832282. PMID 24056411.
- Basile W, Sachenkova O, Light S, Elofsson A (March 2017). "High GC content causes orphan proteins to be intrinsically disordered". PLOS Computational Biology. 13 (3): e1005375. Bibcode:2017PLSCB..13E5375B. doi:10.1371/journal.pcbi.1005375. PMC 5389847. PMID 28355220.
- Bitard-Feildel T, Heberlein M, Bornberg-Bauer E, Callebaut I (December 2015). "Detection of orphan domains in Drosophila using "hydrophobic cluster analysis"". Biochimie. 119: 244–53. doi:10.1016/j.biochi.2015.02.019. PMID 25736992.
- Mukherjee S, Panda A, Ghosh TC (June 2015). "Elucidating evolutionary features and functional implications of orphan genes in Leishmania major". Infection, Genetics and Evolution. 32: 330–7. doi:10.1016/j.meegid.2015.03.031. PMID 25843649.
- Jeon J, Choi J, Lee GW, Park SY, Huh A, Dean RA, et al. (February 2015). "Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae". Scientific Reports. 5: 8567. Bibcode:2015NatSR...5E8567J. doi:10.1038/srep08567. PMC 4338423. PMID 25708804.
- Zhuang X, Yang C, Murphy KR, Cheng CC (February 2019). "Molecular mechanism and history of non-sense to sense evolution of antifreeze glycoprotein gene in northern gadids". Proceedings of the National Academy of Sciences of the United States of America. 116 (10): 4400–4405. doi:10.1073/pnas.1817138116. PMC 6410882. PMID 30765531.
- Reinhardt JA, Wanjiru BM, Brant AT, Saelao P, Begun DJ, Jones CD (2013). "De novo ORFs in Drosophila are important to organismal fitness and evolved rapidly from previously non-coding sequences". PLOS Genetics. 9 (10): e1003860. doi:10.1371/journal.pgen.1003860. PMC 3798262. PMID 24146629.
- Dinger ME, Pang KC, Mercer TR, Mattick JS (November 2008). "Differentiating protein-coding and noncoding RNA: challenges and ambiguities". PLOS Computational Biology. 4 (11): e1000176. Bibcode:2008PLSCB...4E0176D. doi:10.1371/journal.pcbi.1000176. PMC 2518207. PMID 19043537.
- Stewart, Nicholas B.; Rogers, Rebekah L.; Malik, Harmit S. (23 September 2019). "Chromosomal rearrangements as a source of new gene formation in Drosophila yakuba". PLOS Genetics. 15 (9): e1008314. doi:10.1371/journal.pgen.1008314. PMC 6776367. PMID 31545792.
- Swanson WJ, Vacquier VD (February 2002). "The rapid evolution of reproductive proteins". Nature Reviews Genetics. 3 (2): 137–44. doi:10.1038/nrg733. PMID 11836507. S2CID 25696990.
- Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, Glanowski S, Tanenbaum DM, White TJ, Sninsky JJ, Hernandez RD, Civello D, Adams MD, Cargill M, Clark AG (October 2005). "Natural selection on protein-coding genes in the human genome". Nature. 437 (7062): 1153–7. Bibcode:2005Natur.437.1153B. doi:10.1038/nature04240. PMID 16237444. S2CID 4423768.
- Clark NL, Aagaard JE, Swanson WJ (January 2006). "Evolution of reproductive proteins from animals and plants". Reproduction. 131 (1): 11–22. doi:10.1530/rep.1.00357. PMID 16388004.
- Gubala AM, Schmitz JF, Kearns MJ, Vinh TT, Bornberg-Bauer E, Wolfner MF, Findlay GD (May 2017). "The Goddard and Saturn Genes Are Essential for Drosophila Male Fertility and May Have Arisen De Novo". Molecular Biology and Evolution. 34 (5): 1066–1082. doi:10.1093/molbev/msx057. PMC 5400382. PMID 28104747.
- Luis Villanueva-Cañas J, Ruiz-Orera J, Agea MI, Gallo M, Andreu D, Albà MM (July 2017). "New Genes and Functional Innovation in Mammals". Genome Biology and Evolution. 9 (7): 1886–1900. doi:10.1093/gbe/evx136. PMC 5554394. PMID 28854603.
- Schmidt EE (July 1996). "Transcriptional promiscuity in testes". Current Biology. 6 (7): 768–9. doi:10.1016/S0960-9822(02)00589-4. PMID 8805310. S2CID 14318566.
- White-Cooper H, Davidson I (July 2011). "Unique aspects of transcription regulation in male germ cells". Cold Spring Harbor Perspectives in Biology. 3 (7): a002626. doi:10.1101/cshperspect.a002626. PMC 3119912. PMID 21555408.
- Kleene KC (August 2001). "A possible meiotic function of the peculiar patterns of gene expression in mammalian spermatogenic cells". Mechanisms of Development. 106 (1–2): 3–23. doi:10.1016/S0925-4773(01)00413-0. PMID 11472831. S2CID 949694.
- David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM (April 2006). "A high-resolution map of transcription in the yeast genome". Proceedings of the National Academy of Sciences of the United States of America. 103 (14): 5320–5. Bibcode:2006PNAS..103.5320D. doi:10.1073/pnas.0601091103. PMC 1414796. PMID 16569694.
- Tisseur M, Kwapisz M, Morillon A (November 2011). "Pervasive transcription - Lessons from yeast". Biochimie. 93 (11): 1889–96. doi:10.1016/j.biochi.2011.07.001. PMID 21771634.
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M (June 2008). "The transcriptional landscape of the yeast genome defined by RNA sequencing". Science. 320 (5881): 1344–9. Bibcode:2008Sci...320.1344N. doi:10.1126/science.1158441. PMC 2951732. PMID 18451266.
- Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, Rozowsky JS, Gerstein MB, Wahlestedt C, Hayashizaki Y, Carninci P, Gingeras TR, Mattick JS (July 2011). "The reality of pervasive transcription". PLOS Biology. 9 (7): e1000625, discussion e1001102. doi:10.1371/journal.pbio.1000625. PMC 3134446. PMID 21765801.
- Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJ, Jackson SE, et al. (September 2014). "Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes". Cell Reports. 8 (5): 1365–79. doi:10.1016/j.celrep.2014.07.045. PMC 4216110. PMID 25159147.
- Ruiz-Orera J, Verdaguer-Grau P, Villanueva-Cañas JL, Messeguer X, Albà MM (May 2018). "Translation of neutrally evolving peptides provides a basis for de novo gene evolution". Nature Ecology & Evolution. 2 (5): 890–896. doi:10.1038/s41559-018-0506-6. hdl:10230/36048. PMID 29556078. S2CID 4959952.
- Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM (September 2014). "Long non-coding RNAs as a source of new peptides". eLife. 3: e03523. arXiv:1405.4174. Bibcode:2014arXiv1405.4174R. doi:10.7554/eLife.03523. PMC 4359382. PMID 25233276.
- Wilson BA, Masel J (2011). "Putatively noncoding transcripts show extensive association with ribosomes". Genome Biology and Evolution. 3: 1245–52. doi:10.1093/gbe/evr099. PMC 3209793. PMID 21948395.
- Chen, J; Brunner, AD; Cogan, JZ; Nuñez, JK; Fields, AP; Adamson, B; Itzhak, DN; Li, JY; Mann, M; Leonetti, MD; Weissman, JS (6 March 2020). "Pervasive functional translation of noncanonical human open reading frames". Science. 367 (6482): 1140–1146. Bibcode:2020Sci...367.1140C. doi:10.1126/science.aay0262. PMC 7289059. PMID 32139545.
- Keefe AD, Szostak JW (April 2001). "Functional proteins from a random-sequence library". Nature. 410 (6829): 715–8. Bibcode:2001Natur.410..715K. doi:10.1038/35070613. PMC 4476321. PMID 11287961.
- Tretyachenko V, Vymětal J, Bednárová L, Kopecký V, Hofbauerová K, Jindrová H, et al. (November 2017). "Random protein sequences can form defined secondary structures and are well-tolerated in vivo". Scientific Reports. 7 (1): 15449. Bibcode:2017NatSR...715449T. doi:10.1038/s41598-017-15635-8. PMC 5684393. PMID 29133927.
- Wright PE, Dyson HJ (January 2015). "Intrinsically disordered proteins in cellular signalling and regulation". Nature Reviews Molecular Cell Biology. 16 (1): 18–29. doi:10.1038/nrm3920. PMC 4405151. PMID 25531225.
- Neme R, Amador C, Yildirim B, McConnell E, Tautz D (June 2017). "Random sequences are an abundant source of bioactive RNAs or peptides". Nature Ecology & Evolution. 1 (6): 0217. doi:10.1038/s41559-017-0127. PMC 5447804. PMID 28580432.
- Silveira AB, Trontin C, Cortijo S, Barau J, Del Bem LE, Loudet O, et al. (April 2013). "Extensive natural epigenetic variation at a de novo originated gene". PLOS Genetics. 9 (4): e1003437. doi:10.1371/journal.pgen.1003437. PMC 3623765. PMID 23593031.
- Kimmins S, Sassone-Corsi P (March 2005). "Chromatin remodelling and epigenetic features of germ cells". Nature. 434 (7033): 583–9. Bibcode:2005Natur.434..583K. doi:10.1038/nature03368. PMID 15800613. S2CID 4373304.
- Rajon E, Masel J (January 2011). "Evolution of molecular error rates and the consequences for evolvability". Proceedings of the National Academy of Sciences of the United States of America. 108 (3): 1082–7. Bibcode:2011PNAS..108.1082R. doi:10.1073/pnas.1012918108. PMC 3024668. PMID 21199946.
- Masel, Joanna (March 2006). "Cryptic Genetic Variation Is Enriched for Potential Adaptations". Genetics. 172 (3): 1985–1991. doi:10.1534/genetics.105.051649. PMC 1456269. PMID 16387877.
- Willis S, Masel J (September 2018). "Gene Birth Contributes to Structural Disorder Encoded by Overlapping Genes". Genetics. 210 (1): 303–313. doi:10.1534/genetics.118.301249. PMC 6116962. PMID 30026186.
- Giacomelli, Michael G.; Hancock, Adam S.; Masel, Joanna (February 2007). "The Conversion of 3′ UTRs into Coding Regions". Molecular Biology and Evolution. 24 (2): 457–464. doi:10.1093/molbev/msl172. PMC 1808353. PMID 17099057.
- Bornberg-Bauer E, Schmitz J, Heberlein M (October 2015). "Emergence of de novo proteins from 'dark genomic matter' by 'grow slow and moult'". Biochemical Society Transactions. 43 (5): 867–73. doi:10.1042/BST20150089. PMID 26517896.
- Wilder, Jason A.; Hewett, Elizabeth K.; Gansner, Meredith E. (December 2009). "Molecular Evolution of GYPC: Evidence for Recent Structural Innovation and Positive Selection in Humans". Molecular Biology and Evolution. 26 (12): 2679–2687. doi:10.1093/molbev/msp183. PMC 2775107. PMID 19679754.
- Vakhrusheva, Anna A.; Kazanov, Marat D.; Mironov, Andrey A.; Bazykin, Georgii A. (17 November 2010). "Evolution of Prokaryotic Genes by Shift of Stop Codons". Journal of Molecular Evolution. 72 (2): 138–146. doi:10.1007/s00239-010-9408-1. PMID 21082168. S2CID 812377.
- Andreatta, Matthew E.; Levine, Joshua A.; Foy, Scott G.; Guzman, Lynette D.; Kosinski, Luke J.; Cordes, Matthew H.J.; Masel, Joanna (June 2015). "The Recent De Novo Origin of Protein C-Termini". Genome Biology and Evolution. 7 (6): 1686–1701. doi:10.1093/gbe/evv098. PMC 4494051. PMID 26002864.
- Kleppe AS, Bornberg-Bauer E (November 2018). "Robustness by intrinsically disordered C-termini and translational readthrough". Nucleic Acids Research. 46 (19): 10184–10194. doi:10.1093/nar/gky778. PMC 6365619. PMID 30247639.
- Klasberg S, Bitard-Feildel T, Callebaut I, Bornberg-Bauer E (July 2018). "Origins and structural properties of novel and de novo protein domains during insect evolution". The FEBS Journal. 285 (14): 2605–2625. doi:10.1111/febs.14504. PMID 29802682.
- Chen S, Krinsky BH, Long M (September 2013). "New genes as drivers of phenotypic evolution". Nature Reviews Genetics. 14 (9): 645–60. doi:10.1038/nrg3521. PMC 4236023. PMID 23949544.
- Suenaga Y, Islam SM, Alagu J, Kaneko Y, Kato M, Tanaka Y, et al. (January 2014). "NCYM, a Cis-antisense gene of MYCN, encodes a de novo evolved protein that inhibits GSK3β resulting in the stabilization of MYCN in human neuroblastomas". PLOS Genetics. 10 (1): e1003996. doi:10.1371/journal.pgen.1003996. PMC 3879166. PMID 24391509.
- Lin B, White JT, Ferguson C, Bumgarner R, Friedman C, Trask B, et al. (February 2000). "PART-1: a novel human prostate-specific, androgen-regulated gene that maps to chromosome 5q12". Cancer Research. 60 (4): 858–63. PMID 10706094.
- Kang M, Ren M, Li Y, Fu Y, Deng M, Li C (July 2018). "Exosome-mediated transfer of lncRNA PART1 induces gefitinib resistance in esophageal squamous cell carcinoma via functioning as a competing endogenous RNA". Journal of Experimental & Clinical Cancer Research. 37 (1): 171. doi:10.1186/s13046-018-0845-9. PMC 6063009. PMID 30049286.
- Samusik N, Krukovskaya L, Meln I, Shilov E, Kozlov AP (2013). "PBOV1 is a human de novo gene with tumor-specific expression that is associated with a positive clinical outcome of cancer". PLOS ONE. 8 (2): e56162. Bibcode:2013PLoSO...856162S. doi:10.1371/journal.pone.0056162. PMC 3572036. PMID 23418531.
- Guerzoni D, McLysaght A (April 2016). "De Novo Genes Arise at a Slow but Steady Rate along the Primate Lineage and Have Been Subject to Incomplete Lineage Sorting". Genome Biology and Evolution. 8 (4): 1222–32. doi:10.1093/gbe/evw074. PMC 4860702. PMID 27056411.
- Pekarsky Y, Rynditch A, Wieser R, Fonatsch C, Gardiner K (September 1997). "Activation of a novel gene in 3q21 and identification of intergenic fusion transcripts with ecotropic viral insertion site I in leukemia". Cancer Research. 57 (18): 3914–9. PMID 9307271.
- Papamichos SI, Margaritis D, Kotsianidis I (2015). "Adaptive Evolution Coupled with Retrotransposon Exaptation Allowed for the Generation of a Human-Protein-Specific Coding Gene That Promotes Cancer Cell Proliferation and Metastasis in Both Haematological Malignancies and Solid Tumours: The Extraordinary Case of MYEOV Gene". Scientifica. 2015: 984706. doi:10.1155/2015/984706. PMC 4629056. PMID 26568894.
- Kozlov AP (2016). "Expression of evolutionarily novel genes in tumors". Infectious Agents and Cancer. 11: 34. doi:10.1186/s13027-016-0077-6. PMC 4949931. PMID 27437030.
- Li CY, Zhang Y, Wang Z, Zhang Y, Cao C, Zhang PW, et al. (March 2010). "A human-specific de novo protein-coding gene associated with human brain functions". PLOS Computational Biology. 6 (3): e1000734. Bibcode:2010PLSCB...6E0734L. doi:10.1371/journal.pcbi.1000734. PMC 2845654. PMID 20376170.
- Zhang YE, Landback P, Vibranovski MD, Long M (October 2011). "Accelerated recruitment of new brain development genes into the human genome". PLOS Biology. 9 (10): e1001179. doi:10.1371/journal.pbio.1001179. PMC 3196496. PMID 22028629.
- Wang J, Xie G, Singh M, Ghanbarian AT, Raskó T, Szvetnik A, et al. (December 2014). "Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells" (PDF). Nature. 516 (7531): 405–9. Bibcode:2014Natur.516..405W. doi:10.1038/nature13804. PMID 25317556. S2CID 205240839.
- Dolstra H, Fredrix H, Maas F, Coulie PG, Brasseur F, Mensink E, et al. (January 1999). "A human minor histocompatibility antigen specific for B cell acute lymphoblastic leukemia". The Journal of Experimental Medicine. 189 (2): 301–8. doi:10.1084/jem.189.2.301. PMC 2192993. PMID 9892612.
- Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. (January 2009). "InterPro: the integrative protein signature database". Nucleic Acids Research. 37 (Database issue): D211-5. doi:10.1093/nar/gkn785. PMC 2686546. PMID 18940856.
- Murphy DN, McLysaght A (2012). "De novo origin of protein-coding genes in murine rodents". PLOS ONE. 7 (11): e48650. Bibcode:2012PLoSO...748650M. doi:10.1371/journal.pone.0048650. PMC 3504067. PMID 23185269.
- Zhang L, Ren Y, Yang T, Li G, Chen J, Gschwend AR, et al. (April 2019). "Rapid evolution of protein diversity by de novo origination in Oryza". Nature Ecology & Evolution. 3 (4): 679–690. doi:10.1038/s41559-019-0822-5. PMID 30858588. S2CID 73728579.
- Prabh, Neel; Rödelsperger, Christian (July 2019). "Divergence, and Mixed Origin Contribute to the Emergence of Orphan Genes in Nematodes". G3: Genes, Genomes, Genetics. 9 (7): 2277–2286. doi:10.1534/g3.119.400326. PMC 6643871. PMID 31088903.