Deoxycytidine kinase
Deoxycytidine kinase (dCK) is an enzyme which is encoded by the DCK gene in humans.[5] dCK predominantly phosphorylates deoxycytidine (dC) and converts dC into deoxycytidine monophosphate. dCK catalyzes one of the initial steps in the nucleoside salvage pathway[6] and has the potential to phosphorylate other preformed nucleosides, specifically deoxyadenosine (dA) and deoxyguanosine (dG), and convert them into their monophosphate forms.[7] There has been recent biomedical research interest in investigating dCK's potential as a therapeutic target for different types of cancer.[6][7][8]






Structure
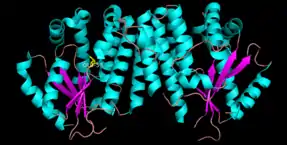
dCK is a homodimer where each monomer subunit consists of multiple alpha helices surrounding a beta sheet core.[9][7][10] Each subunit includes a nucleotide donor binding site, nucleoside acceptor binding site, nucleotide base sensing loop (240-254 residues), insert region (12-15 residues) that connects helices 2 and 3.[9][10] dCK has several different protein conformations but its conformation depends on the nucleoside or nucleotide it binds to. dCK can bind to ADP, ATP, UDP or UTP (phosphoryl group donors) but UDP/UTP binding changes the enzyme's conformation by rearranging the nucleotide base sensing loop as compared to the dCK's conformation when bound to ATP. This change in conformation when a specific phosphoryl donor is bound in the nucleotide binding site determines which nucleoside can bind in the nucleoside binding site.[9][10] For example, it has been observed that when dCK binds to ADP, dCK takes on a "closed" conformation or more compact nucleoside binding site where glutamic acid 53 (Glu53) is brought into closer proximity to directly interact with the nucleoside's 5' hydroxyl group.[9][10]
- One hypothesis for the functionality of the "open" conformation is that the "open" conformation may assist in the initial nucleoside binding and the release of the monophosphate product[9]
Function
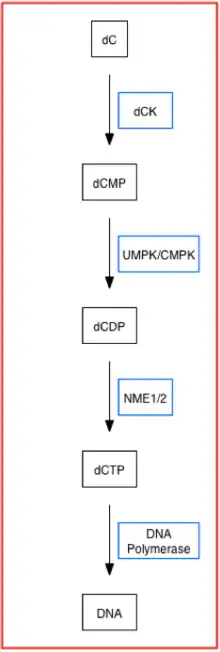
Deoxycytidine kinase (dCK) phosphorylates several deoxyribonucleosides and their nucleoside analogues (a nucleoside with a sugar and a different nucleic acid base substitute or analogue that has unique properties when modified) using phosphate groups from ATP and UTP.[9][10] More specifically, dCK adds the first phosphoryl group to preformed nucleosides and is usually the rate-limiting enzyme of the overall process of converting nucleosides to their deoxynucleoside triphosphate form, or nucleotide form, in the nucleoside salvage pathway.[10] Below is a simplified pathway that displays dCK's role in synthesizing nucleotides using the nucleoside salvage pathway.[8][11]
Glu53 performs base catalysis to deprotonate the hydroxyl group, which allows the now nucleophilic oxygen from the nucleoside 5' hydroxyl group to attack the end of the phosphate chain (gamma phosphate) on the phosphoryl donor (e.g. ATP or UTP). This has deemed the "closed" conformation as the catalytically active conformation since it catalyzes the phosphoryl transfer between phosphoryl donors and receiving nucleosides.[9] Similarly, "open" conformation is generally referred to as the catalytically inactive form since Glu53 is not in close proximity to nucleoside 5' hydroxyl group and will not catalyze the phosphoryl transfer.[9]
Regulation
One method of to regulate both catalytic activity and substrate specificity is a post-translational modification on Serine 74, a residue in the insert region on each of the individual dCK subunits.[9] Although serine 74 is far from dCK's active site, phosphorylation of serine 74 (Ser74) on dCK causes a change in enzyme conformation and influences enzyme kinetics. More specifically, phosphorylation of Ser74 favors dCK to adopt its open (inactive) conformation and allow dCK to become more competent in binding and releasing nucleosides but restricts dCK from transferring phosphoryl groups. dCK's closed (active) conformation allows dCK to transfer phosphoryl groups, but not bind or release nucleosides. The "open" and "closed" states refer to the nucleoside binding site on dCK.[9]
Nucleotide biosynthesis
dCK is a key enzyme in the nucleoside salvage pathway (NSP). More specifically, this pathway recycles preformed nucleosides from degrading DNA molecules to synthesize dNTPs for the cell. The nucleoside salvage pathway can act as an alternative path to produce nucleotides (dNTP's) in case of de novo pathway downregulation.[6] That is, the salvage pathway (and thus dCK) is upregulated when the de novo pathway is downregulated or inhibited in order to compensate for the loss in nucleotide production. Both the de novo pathway (DNP) and the nucleoside salvage pathway (NSP) are anabolic pathways that produce deoxyribonucleotide triphosphates (dNTP's) or nucleotides, the monomers that make up DNA.
Therapeutic implications
Deficiency of dCK is associated with resistance to antiviral and anticancer chemotherapeutic agents. Conversely, increased deoxycytidine kinase activity is associated with increased activation of these agents to cytotoxic nucleoside triphosphate derivatives. dCK is clinically important because of its relationship to drug resistance and sensitivity.[5] Manipulating dCK's enzymatic activity has been shown to have a strong correlation in sensitizing cells to the effects of other drugs (e.g. RNR inhibitors,[6] gemcitabine) or treatments (e.g. ionizing radiation)[11] and so more combination therapies are currently been studied to reduce biological resistance mechanisms and drug tolerance in patients.[6][11][12]
For example, gemcitabine is a FDA-approved pyrimidine nucleoside analogue and a dCK activity based prodrug that has been used to treat pancreatic, breast, bladder and non-small cell lung cancer.[8][11] Mechanistically, dCK, which uptakes preformed nucleosides, adds the first phosphoryl group on dFdC (gemcitabine's original form as a deoxycytidine analog) to convert it into dFdCMP, its monophosphate form.[8][11] Cytidylate kinase or UMP-CMP kinase then adds the second phosphoryl group to form dFdCDP (gemcitabine diphosphate form), which can inhibit ribonucleotide reductase. Nucleoside-diphosphate kinase or nucleoside kinase A adds the third phosphoryl group to form dFdCTP (gemcitabine triphosphate form) which is gemcitabine's active form which inhibits both deoxycytidylate deaminase and DNA polymerase.[8] Although gemcitabine has widely used to treat solid tumors for over a decade, patients taking gemcitabine alone (monotherapy) have been observed to develop chemoresistance to the drug.[8][11]
See also
References
- GRCh38: Ensembl release 89: ENSG00000156136 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000029366 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Entrez Gene: DCK deoxycytidine kinase".
- Nathanson DA, Armijo AL, Tom M, Li Z, Dimitrova E, Austin WR, Nomme J, Campbell DO, Ta L, Le TM, Lee JT, Darvish R, Gordin A, Wei L, Liao HI, Wilks M, Martin C, Sadeghi S, Murphy JM, Boulos N, Phelps ME, Faull KF, Herschman HR, Jung ME, Czernin J, Lavie A, Radu CG (March 2014). "Co-targeting of convergent nucleotide biosynthetic pathways for leukemia eradication". The Journal of Experimental Medicine. 211 (3): 473–86. doi:10.1084/jem.20131738. PMC 3949575. PMID 24567448.
- Sabini E, Ort S, Monnerjahn C, Konrad M, Lavie A (July 2003). "Structure of human dCK suggests strategies to improve anticancer and antiviral therapy". Nature Structural Biology. 10 (7): 513–9. doi:10.1038/nsb942. hdl:11858/00-001M-0000-0012-F0B9-8. PMID 12808445. S2CID 6212685.
- de Sousa Cavalcante L, Monteiro G (October 2014). "Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer". European Journal of Pharmacology. 741: 8–16. doi:10.1016/j.ejphar.2014.07.041. PMID 25084222.
- Hazra S, Szewczak A, Ort S, Konrad M, Lavie A (April 2011). "Post-translational phosphorylation of serine 74 of human deoxycytidine kinase favors the enzyme adopting the open conformation making it competent for nucleoside binding and release". Biochemistry. 50 (14): 2870–80. doi:10.1021/bi2001032. PMC 3071448. PMID 21351740.
- Sabini E, Hazra S, Konrad M, Lavie A (July 2008). "Elucidation of Different Binding Modes of Purine Nucleosides to Human Deoxycytidine Kinase". Journal of Medicinal Chemistry. 51 (14): 4219–25. doi:10.1021/jm800134t. PMC 2636677. PMID 18570408.
- Grégoire V, Rosier JF, De Bast M, Bruniaux M, De Coster B, Octave-Prignot M, Scalliet P (June 2002). "Role of deoxycytidine kinase (dCK) activity in gemcitabine's radioenhancement in mice and human cell lines in vitro". Radiotherapy and Oncology. 63 (3): 329–38. doi:10.1016/s0167-8140(02)00106-8. PMID 12142097.
- Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT, Lipson EJ, Chapman PB, Diaz LA, Vogelstein B, Nowak MA (June 2013). "Evolutionary dynamics of cancer in response to targeted combination therapy". eLife. 2: e00747. doi:10.7554/eLife.00747. PMC 3691570. PMID 23805382.
Further reading
- Hazra S, Szewczak A, Ort S, Konrad M, Lavie A (April 2011). "Post-translational phosphorylation of serine 74 of human deoxycytidine kinase favors the enzyme adopting the open conformation making it competent for nucleoside binding and release". Biochemistry. 50 (14): 2870–80. doi:10.1021/bi2001032. PMC 3071448. PMID 21351740.
- Hazra S, Konrad M, Lavie A (August 2010). "The sugar ring of the nucleoside is required for productive substrate positioning in the active site of human deoxycytidine kinase (dCK): implications for the development of dCK-activated acyclic guanine analogues". Journal of Medicinal Chemistry. 53 (15): 5792–800. doi:10.1021/jm1005379. PMC 2936711. PMID 20684612.
- Hazra S, Ort S, Konrad M, Lavie A (August 2010). "Structural and kinetic characterization of human deoxycytidine kinase variants able to phosphorylate 5-substituted deoxycytidine and thymidine analogues". Biochemistry. 49 (31): 6784–90. doi:10.1021/bi100839e. PMC 2925221. PMID 20614893.
- Hazra S, Sabini E, Ort S, Konrad M, Lavie A (February 2009). "Extending thymidine kinase activity to the catalytic repertoire of human deoxycytidine kinase". Biochemistry. 48 (6): 1256–63. doi:10.1021/bi802062w. PMC 2701478. PMID 19159229.
- Sabini E, Hazra S, Konrad M, Lavie A (July 2008). "Elucidation of different binding modes of purine nucleosides to human deoxycytidine kinase". Journal of Medicinal Chemistry. 51 (14): 4219–25. doi:10.1021/jm800134t. PMC 2636677. PMID 18570408.
- Sabini E, Hazra S, Ort S, Konrad M, Lavie A (May 2008). "Structural basis for substrate promiscuity of dCK". Journal of Molecular Biology. 378 (3): 607–21. doi:10.1016/j.jmb.2008.02.061. PMC 2426910. PMID 18377927.
- McSorley T, Ort S, Hazra S, Lavie A, Konrad M (March 2008). "Mimicking phosphorylation of Ser-74 on human deoxycytidine kinase selectively increases catalytic activity for dC and dC analogues". FEBS Letters. 582 (5): 720–4. doi:10.1016/j.febslet.2008.01.048. PMC 2636680. PMID 18258203.
- Sabini E, Hazra S, Konrad M, Lavie A (June 2007). "Nonenantioselectivity property of human deoxycytidine kinase explained by structures of the enzyme in complex with L- and D-nucleosides". Journal of Medicinal Chemistry. 50 (13): 3004–14. doi:10.1021/jm0700215. PMC 2586175. PMID 17530837.
- Sabini E, Hazra S, Konrad M, Burley SK, Lavie A (2007). "Structural basis for activation of the therapeutic L-nucleoside analogs 3TC and troxacitabine by human deoxycytidine kinase". Nucleic Acids Research. 35 (1): 186–92. doi:10.1093/nar/gkl1038. PMC 1802566. PMID 17158155.
- Arnér ES, Eriksson S (1996). "Mammalian deoxyribonucleoside kinases". Pharmacology & Therapeutics. 67 (2): 155–86. doi:10.1016/0163-7258(95)00015-9. PMID 7494863.
- Chottiner EG, Shewach DS, Datta NS, Ashcraft E, Gribbin D, Ginsburg D, Fox IH, Mitchell BS (February 1991). "Cloning and expression of human deoxycytidine kinase cDNA". Proceedings of the National Academy of Sciences of the United States of America. 88 (4): 1531–5. doi:10.1073/pnas.88.4.1531. PMC 51053. PMID 1996353.
- Eriksson S, Cederlund E, Bergman T, Jörnvall H, Bohman C (March 1991). "Characterization of human deoxycytidine kinase. Correlation with cDNA sequences". FEBS Letters. 280 (2): 363–6. doi:10.1016/0014-5793(91)80332-W. PMID 2013338. S2CID 26841109.
- Yamada Y, Goto H, Ogasawara N (November 1983). "Purine nucleoside kinases in human T- and B-lymphoblasts". Biochimica et Biophysica Acta (BBA) - General Subjects. 761 (1): 34–40. doi:10.1016/0304-4165(83)90359-8. PMID 6315069.
- Hurley MC, Palella TD, Fox IH (December 1983). "Human placental deoxyadenosine and deoxyguanosine phosphorylating activity". The Journal of Biological Chemistry. 258 (24): 15021–7. PMID 6317685.
- Spasokoukotskaja T, Arnér ES, Brosjö O, Gunvén P, Juliusson G, Liliemark J, Eriksson S (1995). "Expression of deoxycytidine kinase and phosphorylation of 2-chlorodeoxyadenosine in human normal and tumour cells and tissues". European Journal of Cancer. 31A (2): 202–8. doi:10.1016/0959-8049(94)00435-8. PMID 7718326.
- Stegmann AP, Honders MW, Bolk MW, Wessels J, Willemze R, Landegent JE (August 1993). "Assignment of the human deoxycytidine kinase (DCK) gene to chromosome 4 band q13.3-q21.1". Genomics. 17 (2): 528–9. doi:10.1006/geno.1993.1365. PMID 8406512.
- Song JJ, Walker S, Chen E, Johnson EE, Spychala J, Gribbin T, Mitchell BS (January 1993). "Genomic structure and chromosomal localization of the human deoxycytidine kinase gene". Proceedings of the National Academy of Sciences of the United States of America. 90 (2): 431–4. doi:10.1073/pnas.90.2.431. PMC 45676. PMID 8421671.
- Johansson M, Brismar S, Karlsson A (October 1997). "Human deoxycytidine kinase is located in the cell nucleus". Proceedings of the National Academy of Sciences of the United States of America. 94 (22): 11941–5. doi:10.1073/pnas.94.22.11941. PMC 23663. PMID 9342341.
- Hatzis P, Al-Madhoon AS, Jüllig M, Petrakis TG, Eriksson S, Talianidis I (November 1998). "The intracellular localization of deoxycytidine kinase". The Journal of Biological Chemistry. 273 (46): 30239–43. doi:10.1074/jbc.273.46.30239. PMID 9804782.
- Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O (November 2001). "Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy". Nature Genetics. 29 (3): 342–4. doi:10.1038/ng751. PMID 11687801. S2CID 3045143.
- Veuger MJ, Heemskerk MH, Honders MW, Willemze R, Barge RM (February 2002). "Functional role of alternatively spliced deoxycytidine kinase in sensitivity to cytarabine of acute myeloid leukemic cells". Blood. 99 (4): 1373–80. doi:10.1182/blood.V99.4.1373. PMID 11830489.
- Innoceta A, Galluzzi L, Ruzzo A, Andreoni F, Chiarantini L, Magnani M (February 2002). "Molecular basis of 2',3'-dideoxycytidine-induced drug resistance in human cells". Molecular and Cellular Biochemistry. 231 (1–2): 173–7. doi:10.1023/A:1014441209108. PMID 11952160. S2CID 11289854.
- Krawiec K, Kierdaszuk B, Shugar D (January 2003). "Inorganic tripolyphosphate (PPP(i)) as a phosphate donor for human deoxyribonucleoside kinases". Biochemical and Biophysical Research Communications. 301 (1): 192–7. doi:10.1016/S0006-291X(02)03007-3. PMID 12535661.
- van der Wilt CL, Kroep JR, Loves WJ, Rots MG, Van Groeningen CJ, Kaspers GJ, Peters GJ (March 2003). "Expression of deoxycytidine kinase in leukaemic cells compared with solid tumour cell lines, liver metastases and normal liver". European Journal of Cancer. 39 (5): 691–7. doi:10.1016/S0959-8049(02)00813-4. PMID 12628850.
- Ge Y, Jensen TL, Matherly LH, Taub JW (December 2003). "Physical and functional interactions between USF and Sp1 proteins regulate human deoxycytidine kinase promoter activity". The Journal of Biological Chemistry. 278 (50): 49901–10. doi:10.1074/jbc.M305085200. PMID 14514691.
- Usova E, Maltseva T, Földesi A, Chattopadhayaya J, Eriksson S (December 2004). "Human deoxycytidine kinase as a deoxyribonucleoside phosphorylase". Journal of Molecular Biology. 344 (5): 1347–58. doi:10.1016/j.jmb.2004.10.016. PMID 15561147.
- Mani RS, Usova EV, Eriksson S, Cass CE (October 2004). "Fluorescence studies of substrate binding to human recombinant deoxycytidine kinase". Nucleosides, Nucleotides & Nucleic Acids. 23 (8–9): 1343–6. doi:10.1081/NCN-200027609. PMID 15571255. S2CID 20686075.
External links
- Deoxycytidine+kinase at the US National Library of Medicine Medical Subject Headings (MeSH)
PDB gallery | |
|---|---|
|








