Ei mechanism
The Ei mechanism (Elimination Internal/Intramolecular), also known as a thermal syn elimination or a pericyclic syn elimination, in organic chemistry is a special type of elimination reaction in which two vicinal substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination.[1] This type of elimination is unique because it is thermally activated and does not require additional reagents unlike regular eliminations which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.
General Features
Compounds that undergo elimination through cyclic transition states upon heating, with no other reagents present, are given the designation as Ei reactions. Depending on the compound, elimination takes place through a four, five, or six-membered transition state.[1][2]
The elimination must be syn and the atoms coplanar for four and five-membered transition states,[3] but coplanarity is not required for six-membered transition states.[1]
There is a substantial amount of evidence to support the existence of the Ei mechanism such as: 1) the kinetics of the reactions were found to be first order,[4] 2) the use of free-radical inhibitors did not affect the rate of the reactions, indicating no free-radical mechanisms are involved [5][6] 3) isotope studies for the Cope elimination indicate the C-H and C-N bonds are partially broken in the transition state,[7] this is also supported by computations that show bond lengthening in the transition state [8] and 4) without the intervention of other mechanisms, the Ei mechanism gives exclusively syn elimination products.
There are many factors that affect the product composition of Ei reactions, but typically they follow Hofmann’s rule and lose a β-hydrogen from the least substituted position, giving the alkene that is less substituted (the opposite of Zaitsev's rule).[1] Some factors affecting product composition include steric effects, conjugation, and stability of the forming alkene.
For acyclic substrates, the Z-isomer is typically the minor product due to the destabilizing gauche interaction in the transition state, but the selectivity is not usually high.[2]
The pyrolysis of N,N-dimethyl-2-phenylcyclohexylamine-N-oxide shows how conformational effects and the stability of the transition state affect product composition for cyclic substrates.[2]

In the trans isomer, there are two cis-β-hydrogens that can eliminate. The major product is the alkene that is in conjugation with the phenyl ring, presumably due to the stabilizing effect on the transition state. In the cis isomer, there is only one cis-B-hydrogen that can eliminate, giving the nonconjugated regioisomer as the major product.
Ester (Acetate) Pyrolysis
The pyrolytic decomposition of esters is an example of a thermal syn elimination. When subjected to temperatures above 400 °C, esters containing β-hydrogens can eliminate a carboxylic acid through a 6-membered transition state, resulting in an alkene.[2][6]
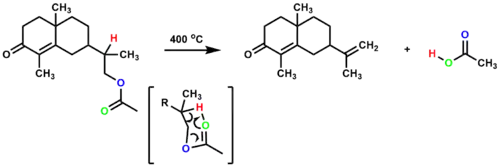
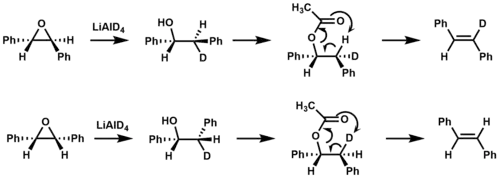
Isotopic labeling was used to confirm that syn elimination occurs during ester pyrolysis in the formation of stilbene.[9]
Sulfur Based
Sulfoxide Elimination
β-hydroxy phenyl sulfoxides were found to undergo thermal elimination through a 5-membered cyclic transition state, yielding β-keto esters and methyl ketones after tautomerization and a sulfenic acid.[10]

Allylic alcohols can be formed from β-hydroxy phenyl sulfoxides that contain a β’-hydrogen through an Ei mechanism, tending to give the β,γ-unsaturation.[11]

1,3-Dienes were found to be formed upon the treatment of an allylic alcohol with an aryl sulfide in the presence of triethylamine.[12] Initially, a sulfenate ester is formed followed by a [2,3]-sigmatropic rearrangement to afford an allylic sulfoxide which undergoes thermal syn elimination to yield the 1,3-diene.

Chugaev Elimination
The Chugaev elimination is the pyrolysis of a xanthate ester, resulting in an olefin.[1][13] To form the xanthate ester, an alcohol reacts with carbon disulfide in the presence of a base, resulting in a metal xanthate which is trapped with an alkylating agent (typically methyl iodide). The olefin is formed through the thermal syn elimination of the β-hydrogen and xanthate ester. The reaction is irreversible because the resulting by-products, carbonyl sulfide and methanethiol, are very stable.
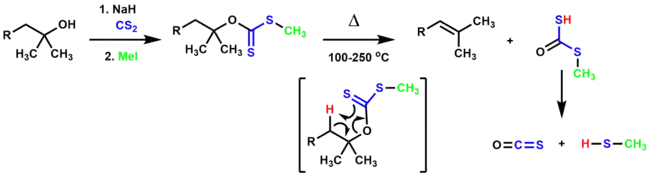
The Chugaev elimination is very similar to the ester pyrolysis, but requires significantly lower temperatures to achieve the elimination, thus making it valuable for rearrangement-prone substrates.
Burgess Dehydration Reaction
The dehydration of secondary and tertiary alcohols to yield an olefin through a sulfamate ester intermediate is called the Burgess dehydration reaction.[13][14][15] The reaction conditions used are typically very mild, giving it some advantage over other dehydration methods for sensitive substrates. This reaction was used during the first total synthesis of taxol to install an exo-methylene group on the C ring.[16]

First, the alcohol displaces the triethylamine on the Burgess reagent, forming the sulfamate ester intermediate. β-hydrogen abstraction and elimination of the sulfamate ester through a 6-membered cyclic transition state yields the alkene.
Thiosulfinate Elimination
Thiosulfinates can eliminate in the manner analogous to sulfoxides. Representative is the fragmentation of allicin to thioacrolein, which will go on to form vinyldithiins. Such reactions are important in the antioxidant chemistry of garlic and other plants of the genus Allium.

Selenium Based
Selenoxide Elimination
The selenoxide elimination has been used in converting ketones, esters, and aldehydes to their α,β-unsaturated derivatives.[1][17]
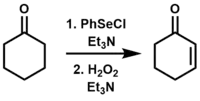
The mechanism for this reaction is analogous to the sulfoxide elimination, which is a thermal syn elimination through a 5-membered cyclic transition state. Selenoxides are preferred for this type of transformation over sulfoxides due to their increased reactivity toward β-elimination, in some cases allowing the elimination to take place at room temperature.[2]

The areneselenic acid generated after the elimination step is in equilibrium with the diphenyl diselenide which can react with olefins to yield β-hydroxy selenides under acidic or neutral conditions. Under basic conditions, this side reaction is suppressed.[18]
Grieco Elimination
The one-pot dehydration of a primary alcohol to give an alkene through an o-nitrophenyl selenoxide intermediate is called the Grieco elimination.[19][20]

The reaction begins with the formation of a selenophosphonium salt which reacts with the alcohol to form an oxaphosphonium salt. The aryl selenium anion displaces tributylphosphine oxide forming the alkyl aryl selenide species. The selenide is then treated with excess hydrogen peroxide leading to the selenoxide which eliminates the β-hydrogen through a 5-member cyclic transition state, yielding an alkene.

The electron-withdrawing nitro group was found to increase both the rate of elimination and the final yield of the olefin.
Nitrogen Based
Cope Elimination
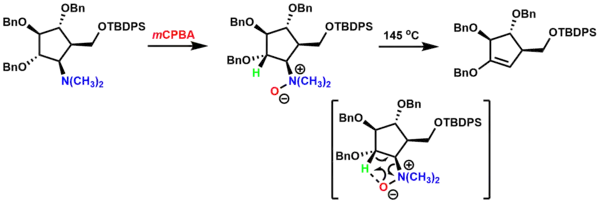
The Cope elimination (Cope reaction) is the elimination of a tertiary amine oxide to yield an alkene and a hydroxylamine through an Ei mechanism.[13][21] The Cope elimination was used in the synthesis of a mannopyranosylamine mimic.[22] The tertiary amine was oxidized to the amine oxide using m-chloroperoxybenzoic acid (mCPBA) and subjected to high temperatures for thermal syn elimination of the β-hydrogen and amine oxide through a cyclic transition state, yielding the alkene. It is worth noting that the indicated hydrogen (in green) is the only hydrogen available for syn elimination.
Cyclic amine oxides (5, 7-10-membered nitrogen containing rings) can also undergo internal syn elimination to yield acyclic hydroxylamines containing terminal alkenes.[13]
Special Cases for the Hofmann Elimination
The mechanism for the Hofmann elimination is generally E2, but can go through an Ei pathway under certain circumstances. For some sterically hindered molecules the base deprotonates a methyl group on the amine instead of the β-hydrogen directly, forming an ylide intermediate which eliminates trimethylamine through a 5-membered transition state, forming the alkene. Deuterium labeling studies confirmed this mechanism by observing the formation of deuterated trimethylamine (and no deuterated water, which would form from the E2 mechanism).[23]

The Wittig modified Hofmann elimination goes through the same Ei mechanism, but instead of using silver oxide and water as base, the Wittig modification uses strong bases like alkylithiums or KNH2/liquid NH3.[24][25]
Iodoso Elimination
Secondary and tertiary alkyl iodides with strongly electron-withdrawing groups at the α-carbon were found to undergo a pericyclic syn elimination when exposed to m-chloroperbenzoic acid (mCPBA).[26] It is proposed that the reaction goes through an iodoso intermediate before the syn elimination of hypoiodous acid.

The scope of this reaction does not include primary alkyl iodides because the iodoso intermediate rearranges to the hypoiodite intermediate, which, under the reaction conditions, is converted to an alcohol. Strongly electron-withdrawing groups suppress the rearrangement pathway, allowing the pericyclic syn elimination pathway to predominate.
References
- March, Jerry (2007). Advanced Organic Chemistry (6th ed.). New York: Wiley. ISBN 0471720917.
- Carey, F. A.; Sunburg, R. J. Advanced Organic Chemistry: Reaction and Synthesis, 5th Ed.; Part B; Springer: New York, 2010
- Branko, J. (1997). "Theoretical studies of thermal syn elimination reaction of organic amine oxide, sulfoxide and phosphoxide by ab initio and density functional methods". Theo. Chem. 389 257-263.
- O’Connor, G.L.; Nace, H. R. (1953). "Further Studies on the Chugaev Reaction and Related Reactions". J. Am. Chem. Soc. 75 2118-.
- Barton, D.H.R.; Head, A.J.; Williams, R.J. (1953). "Stereospecificity in Thermal Elimination Reactions. Part III. The Pyrolysis of (−)-Menthyl Benzoate". J. Chem Soc. 453 1715- .
- Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry, Murdzek, J., Ed. University Science Books, 2006.
- Wright, D.R.; Sims, L.B.; Fry, A. (1983). "Carbon-14 kinetic isotope effects and kinetic studies in the syn-elimination reactions of (2-phenylethyl)dimethylamine oxides". J. Am. Chem. Soc. 105 3714-.
- Kahn, S.D; Erickson, J.A. (1994). "Theoretical Studies of Thermal Syn Elimination Reactions. The Relative Rates of Ethyl Formate, Ethyl Xanthate, and Ethyl Phosphinate Eliminations". J. Am. Chem. Soc. 116 6271-6276.
- Curtin, D.Y.; Kellom, D.B. (1953). "Elimination and Replacement Reactions of dl-erythro- and dl-threo-2-Deutero-1,2-diphenylethanol and Derivatives". J. Am. Chem. Soc. 75 6011-.
- Kinoshita, M.; Kunieda, N.; Nokami, J. (1975). "Pyrolysis of B-Hydroxy Sulfoxides to Ketones". Tetrahedron Lett. 33 2841-2844.
- OKawara, R.; Ueta, K.; Nokami, J. (1978). "Pyrolysis of B-Hydroxy Sulfoxides II. Synthesis of Allylic alcohols". Tetrahedron Lett. 49 4903-4904.
- Wollowitz, S.; Reich, H.J. (1982). "Conversion of Allyl Alcohols to 1,3-Dienes by Sequential Sulfenate-Sulfoxide [2,3] Sigmatropic Rearrangement and Syn Elimination". J. Am. Chem. Soc. 104 7051-7059.
- Kurti, L.; Czako, B. Strategic Applications of Named Reactions in Organic Synthesis, Academic Press, 2005.
- Taylor, E.A.; Penton, H.R.Jr.; Burgess, E.M. (1970). "Synthetic Applications of N-Carboalkoxysulfamate Esters". J. Am. Chem. Soc. 92 5224-5226.
- Taylor, E.A.; Penton, H.R.Jr.; Burgess, E.M. (1973). "Thermal Reactions of Alkyl N-Carbomethoxysulfamate Esters". J. Org. Chem. 38 26-.
- Holton, R.A.; et al. (1994). "First Total Synthesis of Taxol. 2. Completion of the C and D Rings". J. Am. Chem. Soc. 116 1599-1600.
- Lauer, R.F; Young, M.W.; Sharpless, K.B. (1973). "Reactions of Selenoxides: Thermal Syn elimination and H2O-18 Exchange". Tetrahedron Lett. 22 1979-1982.
- Reich, H.J.; Wollowitz, S.; Trend, J.E.; Chow, F.; Wendelborn, D.F. (1978). "Syn Elimination of Alkyl Selenoxides. Side Reactions Involving Selenenic Acids. Structural and Solvent Effects on Rates". J. Org. Chem. 43 1697-.
- Grieco, P.A.; Gilman, S.; Nishizawa, M. (1976). "Organoselenium Chemistry. A Facile One-Step Synthesis of Alkyl Aryl Selenides from Alcohols". J. Org. Chem. 41 1485-.
- Young, M.W.; Sharpless, B.K. (1975). "Olefin Synthesis. Rate Enhancement of the Elimination of Alkyl Aryl Selenoxides by Electron-Withdrawing Substituents". J. Org. Chem. 40 947-.
- Cope, A.C.; Foster, T.T.; Towle, P.H. (1949). "Thermal Decomposition of Amine Oxides to Olefins and Dialkylhydroxylamines". J. Am. Chem. Soc. 71 3929-.
- Vasella, A.; Remen, L. (2002). "Conformationally Biased Mimics of Mannopyranosylamines: Inhibitors of B-Mannosidases?". Helv. Chim. Acta. 85 1118-.
- Cope, A.C.; Mehta, A.S. (1963). "Mechanism of the Hofmann Elimination Reaction: An Ylide Intermediate in the Pyrolysis of a Highly Branched Quaternary Hydroxide". J. Am. Chem. Soc. 85 1949-.
- Wittig, G.; Polster, R. (1957). Ann. Chem. 102 612-.
- Bach, R.D.; Bair, K.W.; Andrzejewski, D. (1972). "The Wittig Modification of the Hofmann Elimination Reaction. Evidence for an alpha’, beta Mechanism". J. Am. Chem. Soc. 94 8608-.
- Reich, H. J., Peake, S. L. (1978). “Hypervalent Organoiodine Chemistry. Syn Elimination of Alkyl Iodoso Compounds”. J. Am. Chem. Soc. 100 4888-.