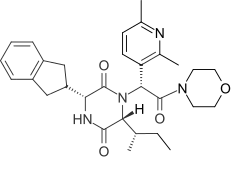
Epelsiban
Epelsiban (INN,[1] USAN,[2] code name GSK-557,296-B) is an orally bioavailable drug which acts as a selective and potent oxytocin receptor antagonist (Ki = 0.13 nM).[3][4] It was initially developed by GlaxoSmithKline (GSK) for the treatment of premature ejaculation in men[5][6] and then as an agent to enhance embryo or blastocyst implantation in women undergoing embryo or blastocyst transfer associated with in vitro fertilization (IVF).,[7] and was also investigated for use in the treatment of adenomyosis.[8]
 | |
| Clinical data | |
|---|---|
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C30H38N4O4 |
| Molar mass | 518.658 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
Discovery and Design
Screening the GSK compound collection and various libraries identified 2,5-diketopiperazines (2,5-DKPs) exemplified by 1 as novel and selective antagonists at the human oxytocin receptor (OTR). The lead, 1, showed potency of Ki = 300nM as a mixture of isomers in the amide side-chain. Initial structure–activity relationship (SAR) studies led to the semi-rigid and chirally pure 2,5-DKP 2 (Ki = 4nM), with cis disposed substituents at C-3 and C-6 and the R side-chain configuration at C-7. The optimal activity was shown to lie in the (3R, 6R, 7R) series (e.g., 2, 3) and an indanyl group was preferred at C-3, while at C-6, a 4-carbon branched alkyl was shown to be optimal.[4]
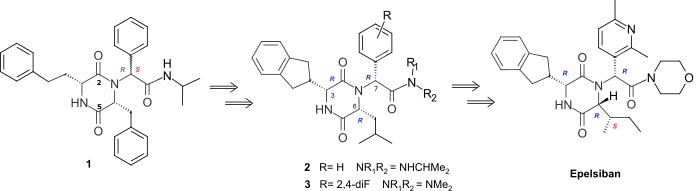
The (3R, 6R, 7R) series 2, 3 showed very good levels of selectivity relative to the vasopressin receptors. However, although all the (3R, 6R, 7R) isomers of the monosubstituted aryl 2,5-DKPs with wide range of different functionality had similarly high levels of potency, they all had low bioavailability in the rat. Optimization of the pharmacokinetic (PK) profile of this template was achieved by property-based design using an estimated of human oral absorption (EHOA) which focused the SAR on 2,5-DKPs with small exocyclic aromatic rings combined with small amides.[4] This resulted in the 2’,4’-difluoro dimethylamide 3 which achieved good oral bioavailability in the rat (53%) and dog (51%) whilst retaining good oxytocin antagonist potency (Ki= 0.63nM) and >1000 fold selectivity relative to the human vasopressin V1A, V2, V1B receptors. The introduction of polar hetereocycles to improve the solubility and human cytochrome P450 (Cyp450) enzyme profile of 3 using intrinsic clearance in microsomes to drive the improvements in the later’s pharmacokinetic profile lead to the 2’,6’-dimethyl-3’-pyridyl morpholine amide Epelsiban.
Mechanism of action
In-vitro binding inhibition data showed that Epelsiban is a highly potent and selective non-peptide oxytocin antagonist with sub-namomolar potency at the human oxytocin receptor (hOTR) Ki = 0.13 nM and with>50000-fold, >63000-fold, and >31000-fold selectivity over the human V1a, V1b and V2 vasopressin receptors. It is also 100-fold more potent at the hOTR than atosiban (a marketed intravenous peptide oxytocin antagonist) and is 5-fold more potent against the hOTR, and more selective against the human vasopressin receptors, especially V2, than retosiban. High in vivo oxytocin antagonist potency was demonstrated in the anesthetized rat model, where uterine contractions were elicited by intravenous administration of oxytocin and reduction in uterine contractility was measured after subsequent intravenous administrations of increasing doses of Epelsiban, which gave an IC50 of 192nM.
Pharmacokinetics
Epelsiban has a good Cyp450 profile with no significant inhibition IC50 > 100μM together with no time-dependent inhibition observed against the five Cyp450 isozymes (1A2, 2C9, 2C19. 2D6, 3A4 DEF, 3A4 7BQ). In addition, Epelsiban has low intrinsic clearance in all four species (rat, dog. cyno monkey, human), a good PK profile in the rat with a bioavailability of 55%, oral exposure and bioavailability in the cynomolgus monkey comparable to retosiban, and good aqueous solubility (33 mg/ml as the besylate salt).
Pharmacology
Epelsiban was investigated for a potential role in benign prostatic hyperplasia also called prostate enlargement.[9] Oxytocin treatment induces prostate enlargement in mice and produces contractions of the prostate through its specific receptor.[10] Oxytocin concentrations are elevated in prostatic tissue from patients with benign prostatic hyperplasia. Epelsiban was found to inhibit the contractile effect of oxytocin in human prostatic tissue through its specific oxytocin receptors in a concentration-dependent manner.[9] suggesting a potential role in the treatment of benign prostatic hyperplasia. The selective antagonist Epelsiban was designed to work on peripheral human oxytocin receptors and not to readily pass the blood-brain barrier.[4] However Epelsiban was found to inhibited brain oxytocin receptors mediating ejaculation, when given intraventricularly to rodents.[5] As expected, despite this success achieved in mice, oral epelsiban in humans at 50 or 150 mg has not shown satisfactory results in a double blind, placebo-controlled trial.[6] This suggested that a central nervous system (CNS) penetrant oxytocin receptor antagonist would show an effect on ejaculation when given systemically. This has been achieved with the Cligosiban which has good CNS penetration.[11] Epelsiban was also investigated as an agent to enhance embryo or blastocyst implantation in women undergoing embryo or blastocyst transfer associated with in vitro fertilization (IVF).[7] and for use in the treatment of adenomyosis.[8] However the development of epelsiban for adenomyosis was terminated in December 2016 for strategic reasons and not because of any safety concerns.
Synthesis
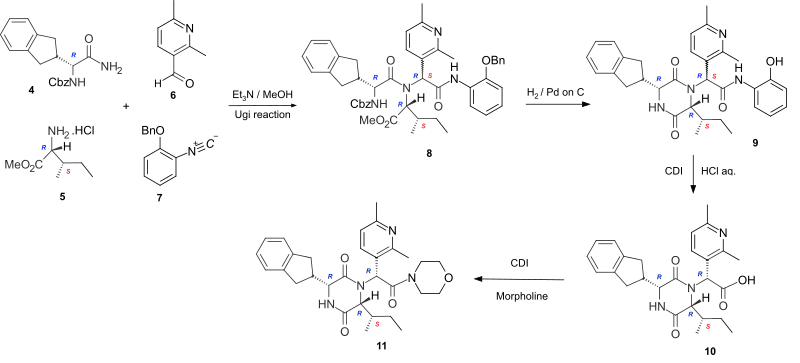
The cyclic dipeptide Epelsiban is formed by cyclizing the corresponding linear dipeptide. In the highly stereoselective synthesis of Epelsiban 11, the linear peptide 8 is formed by the four-component Ugi reaction of the carboxybenzyl (Cbz) protected R-indanylglycine 4, D-alloisoleucine methyl ester hydrochloride 5, 2,6-dimethylpyridine-3-carboxaldehyde 6 and 2-benzyloxyphenylisonitrile 7. Hydrogenation to remove the Cbz and benzyl protecting groups, enabled cyclization of the linear peptide 8 to occur to give the phenolic cyclic dipeptide 9. Hydrolysis of the phenolic amide, by reaction with carbonyl diimidazole (CDI), followed by the addition of aqueous hydrochloric acid gave the acid 10 which was converted to the amide Epelsiban 11 by activating the acid with the peptide coupling reagent CDI, followed by the addition of morpholine.[3] In this short lab-scale synthesis although the linear peptide 8 and the cyclic dipeptide 9 are a mixture of diastereoisomers (7RS) at the exocyclic amide, the hydrochloric acid hydrolysis of the activated phenolic amide caused epimerisation at the exocyclic position and yielded the acid 10 with the required (7R)-stereochemistry as the major product.
History
In screening the GSK compound collection and various libraries, a key consideration was to choose a template with good levels of selectivity over the three vasopressin receptors which are structurally similar to the oxytocin receptor. In addition all templates were also assessed by in silico profiling and suitable templates were evaluated in vitro for predicted CNS penetration. This was to decrease the risk that templates would be chosen that would cross the blood brain barrier and thus block the central effects of oxytocin both in the foetus and in the mother. This identified the small, conformationally constrained, homochiral 2,5-DKP scaffold as the preferred template and lead to the success in designing and developing the highly potent and selective, orally active, peripheral oxytocin antagonist Epelsiban as a clinical candidate.
References
- "International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names (Rec. INN): List 67" (PDF). World Health Organization. p. 62. Retrieved 4 October 2016.
- USAN Council (2011). "Statement on a Nonproprietary Name Adopted by the USAN Council" (PDF). Retrieved 2011-10-28.
- Borthwick AD, Liddle J, Davies DE, Exall AM, Hamlett C, Hickey DM, Mason AM, Smith IE, Nerozzi F, Peace S, Pollard D, Sollis SL, Allen MJ, Woollard PM, Pullen MA, Westfall TD, Stanislaus DJ (January 2012). "Pyridyl-2,5-diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists: synthesis, pharmacokinetics, and in vivo potency". Journal of Medicinal Chemistry. 55 (2): 783–96. doi:10.1021/jm201287w. PMID 22239250.
- Borthwick AD, Liddle J (January 2013). "Retosiban and Epelsiban: Potent and Selective Orally available Oxytocin Antagonists". In Domling A (ed.). Methods and Principles in Medicinal Chemistry: Protein-Protein Interactions in Drug Discovery. Weinheim: Wiley-VCH. pp. 225–256. doi:10.1002/9783527648207.ch10. ISBN 978-3-527-33107-9.
- Clément P, Bernabé J, Compagnie S, Alexandre L, McCallum S, Giuliano F (August 2013). "Inhibition of ejaculation by the non‐peptide oxytocin receptor antagonist GSK 557296: a multi‐level site of action". British Journal of Pharmacology. 169 (7): 1477–1485. doi:10.1111/bph.12198. PMC 3724105. PMID 23530818.
- Shinghal R, Barnes A, Mahar KM, Stier B, Giancaterino L, Condreay LD, Black L, McCallum SW (October 2013). "Safety and efficacy of epelsiban in the treatment of men with premature ejaculation: A randomized, double‐blind, placebo‐controlled, fixed‐dose study". The Journal of Sexual Medicine. 10 (10): 2506–2517. doi:10.1111/jsm.12272. PMID 23937679.
- Mahar KM, Stier B, Fries M, McCallum SW (November 2015). "A single- and multiple-dose study to investigate the pharmacokinetics of epelsiban and its metabolite, GSK2395448, in healthy female volunteers". Clinical Pharmacology in Drug Development. 4 (6): 418–426. doi:10.1002/cpdd.210. PMID 27137713. S2CID 23903528.
- Mahar KM, Enslin MB, Gress A, Amrine-Madsen H, Cooper M (January 2018). "Single‐and Multiple‐Day Dosing Studies to Investigate High‐Dose Pharmacokinetics of Epelsiban and Its Metabolite, GSK2395448, in Healthy Female Volunteers". Clinical Pharmacology in Drug Development. 7 (1): 33–43. doi:10.1002/cpdd.363. PMID 28556598. S2CID 41083332.
- Rouget C, Rekik M, Camparo P, Botto H, Rischmann P, Lluel P, Palea S, McCallum SW, Westfall TD (April 2012). "1568 Oxytocin Produces Contraction of Human Isolated Prostate, an Effect Blocked by the Novel and Selective Oxytocin Receptor Antagonist Gsk557296 Potential Role in Benign Prostatic Hyperplasia". The Journal of Urology. 187 (4S): e635. doi:10.1016/j.juro.2012.02.1340.
- Xu H, Fu S, Chen Y, Chen Q, Gu M, Liu C, Qiao Z, Zhou J, Wang Z (April 2017). "Oxytocin: its role in benign prostatic hyperplasia via the ERK pathway". Clinical Science. 131 (7): 595–607. doi:10.1042/CS20170030. PMID 28130436.
- Wayman C, Russell R, Tang K, Weibly L, Gaboardi S, Fisher L, Allers K, Jackson M, Hawcock T, Robinson N, Wilson L (December 2018). "Cligosiban, a novel brain-penetrant, selective oxytocin receptor antagonist, inhibits ejaculatory physiology in rodents". The Journal of Sexual Medicine. 15 (12): 1698–7. doi:10.1016/j.jsxm.2018.10.008. PMID 30527053.