Molecular neuroscience
Molecular neuroscience is a branch of neuroscience that observes concepts in molecular biology applied to the nervous systems of animals. The scope of this subject covers topics such as molecular neuroanatomy, mechanisms of molecular signaling in the nervous system, the effects of genetics and epigenetics on neuronal development, and the molecular basis for neuroplasticity and neurodegenerative diseases.[1] As with molecular biology, molecular neuroscience is a relatively new field that is considerably dynamic.
Locating neurotransmitters
In molecular biology, communication between neurons typically occurs by chemical transmission across gaps between the cells called synapses. The transmitted chemicals, known as neurotransmitters, regulate a significant fraction of vital body functions.[2] It is possible to anatomically locate neurotransmitters by labeling techniques. It is possible to chemically identify certain neurotransmitters such as catecholamines by fixing neural tissue sections with formaldehyde. This can give rise to formaldehyde-induced fluorescence when exposed to ultraviolet light. Dopamine, a catecholamine, was identified in the nematode C. elegans by using this technique.[3] Immunocytochemistry, which involves raising antibodies against targeted chemical or biological entities, includes a few other techniques of interest. A targeted neurotransmitter could be specifically tagged by primary and secondary antibodies with radioactive labeling in order to identify the neurotransmitter by autoradiography. The presence of neurotransmitters (though not necessarily the location) can be observed in enzyme-linked immunocytochemistry or enzyme--linked immunosorbent assays (ELISA) in which substrate-binding in the enzymatic assays can induce precipitates, fluorophores, or chemiluminescence. In the event that neurotransmitters cannot be histochemically identified, an alternative method is to locate them by their neural uptake mechanisms.[1]
Voltage-gated ion channels
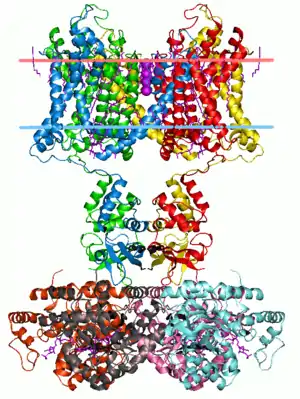
Excitable cells in living organisms have voltage-gated ion channels. These can be observed throughout the nervous system in neurons. The first ion channels to be characterized were the sodium and potassium ion channels by A.L. Hodgkin and A.F. Huxley in the 1950s upon studying the giant axon of the squid genus Loligo. Their research demonstrated the selective permeability of cellular membranes, dependent on physiological conditions, and the electrical effects that result from these permeabilities to produce action potentials.[4]
Sodium ion channels
Sodium channels were the first voltage-gated ion channels to be isolated in 1984 from the eel Electrophorus electricus by Shosaku Numa. The pufferfish toxin tetrodotoxin (TTX), a sodium channel blocker, was used to isolate the sodium channel protein by binding it using the column chromatography technique for chemical separation. The amino acid sequence of the protein was analyzed by Edman degradation and then used to construct a cDNA library which could be used to clone the channel protein. Cloning the channel itself allowed for applications such as identifying the same channels in other animals.[1] Sodium channels are known for working in concert with potassium channels during the development of graded potentials and action potentials. Sodium channels allow an influx of Na+ ions into a neuron, resulting in a depolarization from the resting membrane potential of a neuron to lead to a graded potential or action potential, depending on the degree of depolarization.[5]
Potassium ion channels
Potassium channels come in a variety of forms, are present in most eukaryotic cells, and typically tend to stabilize the cell membrane at the potassium equilibrium potential. As with sodium ions, graded potentials and action potentials are also dependent on potassium channels. While influx of Na+ ions into a neuron induce cellular depolarization, efflux of K+ ions out of a neuron causes a cell to repolarize to resting membrane potential. The activation of potassium ion channels themselves are dependent on the depolarization resulting from Na+ influx during an action potential.[1] As with sodium channels, the potassium channels have their own toxins that block channel protein action. An example of such a toxin is the large cation, tetraethylammonium (TEA), but it is notable that the toxin does not have the same mechanism of action on all potassium channels, given the variety of channel types across species. The presence of potassium channels was first identified in Drosophila melanogaster mutant flies that shook uncontrollably upon anesthesia due to problems in cellular repolarization that led to abnormal neuron and muscle electrophysiology. Potassium channels were first identified by manipulating molecular genetics (of the flies) instead of performing channel protein purification because there were no known high-affinity ligands for potassium channels (such as TEA) at the time of discovery.[1][6]
Calcium ion channels
Calcium channels are important for certain cell-signaling cascades as well as neurotransmitter release at axon terminals. A variety of different types of calcium ion channels are found in excitable cells. As with sodium ion channels, calcium ion channels have been isolated and cloned by chromatographic purification techniques. It is notable, as with the case of neurotransmitter release, that calcium channels can interact with intracellular proteins and plays a strong role in signaling, especially in locations such as the sarcoplasmic reticulum of muscle cells.[1]
Receptors
Various types of receptors can be used for cell signaling and communication and can include ionotropic receptors and metabotropic receptors. These cell surface receptor types are differentiated by the mechanism and duration of action with ionotropic receptors being associated with fast signal transmission and metabotropic receptors being associated with slow signal transmission. Metabotropic receptors happen to cover a wide variety of cell-surface receptors with notably different signaling cascades.[1][5]
Ionotropic receptors
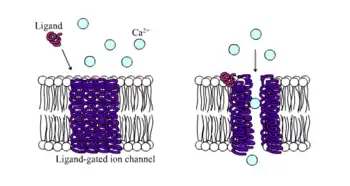
Ionotropic receptors, otherwise known as ligand-gated ion channels, are fast acting receptors that mediate neural and physiological function by ion channel flow with ligand-binding. Nicotinic, GABA, and Glutamate receptors are among some of the cell surface receptors regulated by ligand-gated ion channel flow. GABA is the brain's main inhibitory neurotransmitter and glutamate is the brain's main excitatory neurotransmitter.[1]
GABA receptors
GABAA and GABAC receptors are known to be ionotropic, while the GABAB receptor is metabotropic. GABAA receptors mediate fast inhibitory responses in the central nervous system (CNS) and are found on neurons, glial cells, and adrenal medulla cells. It is responsible for inducing Cl− ion influx into cells, thereby reducing the probability that membrane depolarization will occur upon the arrival of a graded potential or an action potential. GABA receptors can also interact with non-endogenous ligands to influence activity. For example, the compound diazepam (marketed as Valium) is an allosteric agonist which increases the affinity of the receptor for GABA. The increased physiological inhibitory effects resulting from increased GABA binding make diazepam a useful tranquilizer or anticonvulsant (antiepileptic drugs). On the other hand, GABA receptors can also be targeted by decreasing Cl− cellular influx with the effect of convulsants like picrotoxin. The antagonistic mechanism of action for this compound is not directly on the GABA receptor, but there are other compounds that are capable of allosteric inactivation, including T-butylbicyclophorothionate (TBPS) and pentylenetetrazole (PZT). Compared with GABAA, GABAC receptors have a higher affinity for GABA, they are likely to be longer-lasting in activity, and their responses are likely to be generated by lower GABA concentrations.[1]
Glutamate receptors
Ionotropic glutamate receptors can include NMDA, AMPA, and kainate receptors. These receptors are named after agonists that facilitate glutamate activity. NMDA receptors are notable for their excitatory mechanisms to affect neuronal plasticity in learning and memory, as well as neuropathologies such as stroke and epilepsy. NDMA receptors have multiple binding sites just like ionotropic GABA receptors and can be influenced by co-agonists such the glycine neurotransmitter or phencyclidine (PCP). The NMDA receptors carry a current by Ca2+ ions and can be blocked by extracellular Mg2+ ions depending on voltage and membrane potential. This Ca2+ influx is increased by excitatory postsynaptic potentials (EPSPs) produced by NMDA receptors, activating Ca2+-based signaling cascades (such as neurotransmitter release). AMPA generate shorter and larger excitatory postsynaptic currents than other ionotropic glutamate receptors.[5]
Nicotinic ACh receptors
Nicotinic receptors bind the acetylcholine (ACh) neurotransmitter to produce non-selective cation channel flow that generates excitatory postsynaptic responses. Receptor activity, which can be influenced by nicotine consumption, produces feelings of euphoria, relaxation, and inevitably addiction in high levels.[5]
Metabotropic receptors
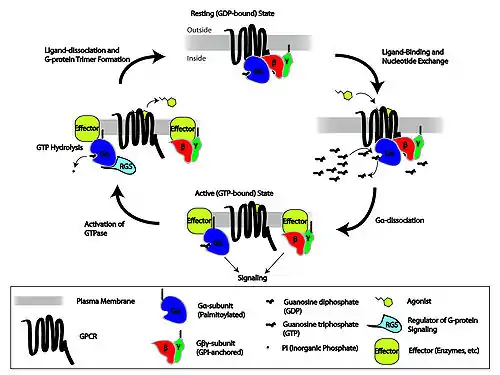
Metabotropic receptors, are slow response receptors in postsynaptic cells. Typically these slow responses are characterized by more elaborate intracellular changes in biochemistry. Responses of neurotransmitter uptake by metabotropic receptors can result in the activation of intracellualar enzymes and cascades involving second messengers, as is the case with G protein-linked receptors. Various metabotropic receptors can include certain glutamate receptors, muscarinic ACh receptors, GABAB receptors, and receptor tyrosine kinases.
G protein-linked receptors
The G protein-linked signaling cascade can significantly amplify the signal of a particular neurotransmitter to produce hundreds to thousands of second messengers in a cell. The mechanism of action by which G protein-linked receptors cause a signaling cascade is as follows:
- Neurotransmitter binds to the receptor
- The receptor undergoes a conformational change to allow G-protein complex binding
- GDP is exchanged with GTP upon G protein complex binding to the receptor
- The α-subunit of the G protein complex is bound to GTP and separates to bind with a target protein such as adenylate cyclase
- The binding to the target protein either increases or decreases the rate of second messenger (such as cyclic AMP) production
- GTPase hydrolyzes the α-subunit so that is bound to GDP and the α-subunit returns to the G protein complex inactive
Neurotransmitter release
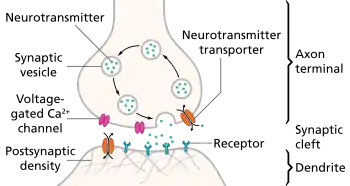
Neurotransmitters are released in discrete packets known as quanta from the axon terminal of one neuron to the dendrites of another across a synapse. These quanta have been identified by electron microscopy as synaptic vesicles. Two types of vesicles are small synaptic vessicles (SSVs), which are about 40-60nm in diameter, and large dense-core vesicles (LDCVs), electron-dense vesicles approximately 120-200nm in diameter.[1] The former is derived from endosomes and houses neurotransmitters such as acetylcholine, glutamate, GABA, and glycine. The latter is derived from the Golgi apparatus and houses larger neurotransmitters such as catecholamines and other peptide neurotransmitters.[7] Neurotransmitters are released from an axon terminal and bind to postsynaptic dendrites in the following procession:[5]
- Mobilization/recruitment of synaptic vesicle from cytoskeleton
- Docking of vesicle (binding) to presynaptic membrane
- Priming of vesicle by ATP (relatively slow step)
- Fusion of primed vesicle with presynaptic membrane and exocytosis of the housed neurotransmitter
- Uptake of neurotransmitters in receptors of a postsynaptic cell
- Initiation or inhibition of action potential in postsynaptic cell depending on whether the neurotransmitters are excitatory or inhibitory (excitatory will result in depolarization of the postsynaptic membrane)
Neurotransmitter release is calcium-dependent
Neurotransmitter release is dependent on an external supply of Ca2+ ions which enter axon terminals via voltage-gated calcium channels. Vesicular fusion with the terminal membrane and release of the neurotransmitter is caused by the generation of Ca2+ gradients induced by incoming action potentials. The Ca2+ ions cause the mobilization of newly synthesized vesicles from a reserve pool to undergo this membrane fusion. This mechanism of action was discovered in squid giant axons.[8] Lowering intracellular Ca2+ ions provides a direct inhibitory effect on neurotransmitter release.[1] After release of the neurotransmitter occurs, vesicular membranes are recycled to their origins of production. Calcium ion channels can vary depending on the location of incidence. For example, the channels at an axon terminal differ from the typical calcium channels of a cell body (whether neural or not). Even at axon terminals, calcium ion channel types can vary, as is the case with P-type calcium channels located at the neuromuscular junction.[1]
Neuronal gene expression
Sex differences
Differences in sex determination are controlled by sex chromosomes. Sex hormonal releases have a significant effect on sexual dimorphisms (phenotypic differentiation of sexual characteristics) of the brain. Recent studies seem to suggest that regulating these dimorphisms has implications for understanding normal and abnormal brain function. Sexual dimorphisms may be significantly influenced by sex-based brain gene expression which varies from species to species.
Animal models such as rodents, Drosophila melanogaster, and Caenorhabditis elegans, have been used to observe the origins and/or extent of sex bias in the brain versus the hormone-producing gonads of an animal. With the rodents, studies on genetic manipulation of sex chromosomes resulted in an effect on one sex that was completely opposite of the effect in the other sex. For example, a knockout of a particular gene only resulted in anxiety-like effects in males. With studies on D. menlanogaster it was found that a large brain sex bias of expression occurred even after the gonads were removed, suggesting that sex bias could be independent of hormonal control in certain aspects.[9]
Observing sex-biased genes has the potential for clinical significance in observing brain physiology and the potential for related (whether directly or indirectly) neurological disorders. Examples of diseases with sex biases in development include Huntington's disease, cerebral ischemia, and Alzheimer's disease.[9]
Epigenetics of the brain
Many brain functions can be influenced at the cellular and molecular level by variations and changes in gene expression, without altering the sequence of DNA in an organism. This is otherwise known as epigenetic regulation. Examples of epigenetic mechanisms include histone modifications and DNA methylation. Such changes have been found to be strongly influential in the incidence of brain disease, mental illness, and addiction.[10] Epigenetic control has been shown to be involved in high levels of plasticity in early development, thereby defining its importance in the critical period of an organism.[11] Examples of how epigenetic changes can affect the human brain are as follows:
- Higher methylation levels in rRNA genes in the hippocampus of the brain results in a lower production of proteins and thus limited hippocampal function can result in learning and memory impairment and resultant suicidal tendencies.[12]
- In a study comparing genetic differences between healthy people and psychiatric patients 60 different epigenetic markers associated with brain cell signaling were found.[12]
- Environmental factors such as child abuse appears to cause the expression of an epigenetic tag on glucocorticoid receptors (associated with stress responses) that was not found in suicide victims.[12] This is an example of experience-dependent plasticity.
- Environmental enrichment in individuals is associated with increased hippocampal gene histone acetylation and thus improved memory consolidation (notably spatial memory).[11]
Molecular mechanisms of neurodegenerative diseases
Excitotoxicity and glutamate receptors
Excitotoxicity is phenomenon in which glutamate receptors are inappropriately activated. It can be caused by prolonged excitatory synaptic transmission in which high levels of glutamate neurotransmitter cause excessive activation in a postsynaptic neuron that can result in the death of the postsynaptic neuron. Following brain injury (such as from ischemia), it has been found that excitotoxicity is a significant cause of neuronal damage. This can be understandable in the case where sudden perfusion of blood after reduced blood flow to the brain can result in excessive synaptic activity caused by the presence of increased glutamate and aspartate during the period of ischemia.[5][13]
Alzheimer's disease
Alzheimer's disease is the most common neurodegenerative disease and is the most common form of dementia in the elderly. The disorder is characterized by progressive loss of memory and various cognitive functions. It is hypothesized that the deposition of amyloid-β peptide (40-42 amino acid residues) in the brain is integral in the incidence of Alzheimer's disease. Accumulation is purported to block hippocampal long-term potentiation. It is also possible that a receptor for amyloid-β oligomers could be a prion protein.[14]
Parkinson's disease
Parkinson's disease is the second most common neurodegenerative disease after Alzheimer's disease. It is a hypokinetic movement basal ganglia disease caused by the loss of dopaminergic neurons in the substantia nigra of the human brain. The inhibitory outflow of the basal ganglia is thus not decreased, and so upper motor neurons, mediated by the thalamus, are not activated in a timely manner. Specific symptoms include rigidity, postural problems, slow movements, and tremors. Blocking GABA receptor input from medium spiny neurons to reticulata cells, causes inhibition of upper motor neurons similar to the inhibition that occurs in Parkinson's disease.[5]
Huntington's disease
Huntington's disease is a hyperkinetic movement basal ganglia disease caused by lack of normal inhibitory inputs from medium spiny neurons of the basal ganglia. This poses the opposite effects of those associated with Parkinson's disease, including inappropriate activation of upper motor neurons. As with the GABAergic mechanisms observed in relation to Parkinson's disease, a GABA agonist injected into the substantia nigra pars reticulata decreases inhibition of upper motor neurons, resulting in ballistic involuntary motor movements, similar to symptoms of Huntington's disease.[5]
References
- Longstaff, Alan; Revest, Patricia (1998). Molecular Neuroscience. Garland Science. ISBN 978-1859962503.
- "What are Neurotransmitters?". Archived from the original on 25 September 2019. Retrieved 1 November 2013.
- Riddle, Donald (1998). C. Elegans II. New York: Cold Spring Harbor Laboratory Press. ISBN 978-0879695323.
- Hodgkin, Allan L.; Andrew F. Huxley (1952). "The dual effect of membrane potential on sodium conductance in the giant axon of Loligo" (PDF). The Journal of Physiology. 116 (4): 497–506. doi:10.1113/jphysiol.1952.sp004719. PMC 1392212. PMID 14946715.
- Purves, Dale (2012). Neuroscience (5th ed.). Massachusetts, USA: Sinauer Associates, Inc. p. 80. ISBN 978-0-87893-695-3.
- Kamb, Alexander; Linda E. Iverson; Mark A. Tanouye (31 July 1987). "Molecular characterization of Shaker, a Drosophila gene that encodes a potassium channel". Cell. 50 (3): 405–413. doi:10.1016/0092-8674(87)90494-6. PMID 2440582.
- Davies, R. Wayne; Brian J. Morris (1997). Molecular Biology of the Neuron. Oxford, UK: BIOS Scientific Publishers Ltd. ISBN 978-1859962404.
- Dipolo, R.; C. Caputo; F. Bezanilla (March 1983). "Voltage-dependent calcium channel in the squid axon". Proc Natl Acad Sci U S A. 80 (6): 1743–1745. Bibcode:1983PNAS...80.1743D. doi:10.1073/pnas.80.6.1743. PMC 393680. PMID 6300873.
- Jazin, E.; Cahill, L. (January 2010). "Sex differences in molecular neuroscience: from fruit flies to humans". Nature Reviews Neuroscience. 11 (1): 9–17. doi:10.1038/nrn2754. PMID 20019686.
- "Epigenetics and the Human Brain". Genetics Science and Learning Center at The University of Utah. Retrieved 10 November 2013.
- Fagiolini, Michela; Catherin L. Jensen; Frances A. Champagne (2009). "Epigenetic Influences on brain development and plasticity" (PDF). Current Opinion in Neurobiology. 19 (2): 1–6. doi:10.1016/j.conb.2009.05.009. PMC 2745597. PMID 19545993. Archived from the original (PDF) on 22 June 2010. Retrieved 14 November 2013.
- Tsankova, Nadia; Renthal, William; Kumar, Arvind; Nestler, Eric J. (2007). "Epigenetic Regulation in Psychiatric Disorders". Nature Reviews Neuroscience. 8 (5): 355–367. doi:10.1038/nrn2132. PMID 17453016.
- Lau, A.; M. Tymianski (2010). "Glutamate receptors, neurotoxicity and neurodegeneration". Pflügers Arch. 460 (2): 525–542. doi:10.1007/s00424-010-0809-1. PMID 20229265.
- Laren, Juha; David A. Gimbel; Haakon B. Nygaard; John W. Gilbert (February 2009). "Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers". Nature. 457 (7233): 1128–1132. Bibcode:2009Natur.457.1128L. doi:10.1038/nature07761. PMC 2748841. PMID 19242475.