PASLI disease
PASLI disease is a rare genetic disorder of the immune system. PASLI stands for “p110 delta activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency.” The immunodeficiency manifests as recurrent infections usually starting in childhood. These include bacterial infections of the respiratory system and chronic viremia due to Epstein-Barr virus (EBV) and/or cytomegalovirus (CMV). Individuals with PASLI disease also have an increased risk of EBV-associated lymphoma. Investigators Carrie Lucas, Michael Lenardo, and Gulbu Uzel at the National Institute of Allergy and Infectious Diseases at the U.S. National Institutes of Health and Sergey Nejentsev at the University of Cambridge, UK simultaneously described a mutation causing this condition which they called Activated PI3K Delta Syndrome (APDS).[1][2]
| PASLI disease | |
|---|---|
| Specialty | Medical genetics |
Signs and symptoms
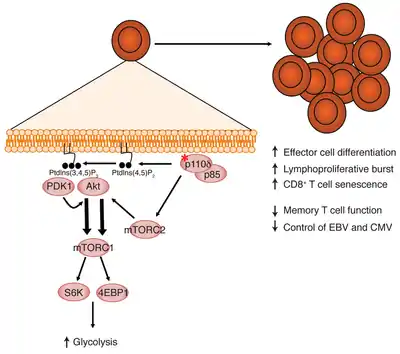
Clinically, PASLI disease is characterized by recurrent sinopulmonary infections that can lead to progressive airway damage. People also have lymphoproliferation (large lymph nodes and spleen), chronic viremia due to EBV or CMV, distinctive lymphoid nodules at mucosal surfaces, autoimmune cytopenias, and EBV-driven B cell lymphoma. Importantly, the clinical presentations and disease courses are variable with some individuals severely affected, whereas others show little manifestation of disease. This “variable expressivity,” even within the same family, can be striking and may be explained by differences in lifestyle, exposure to pathogens, treatment efficacy, or other genetic modifiers.[1][2]
Genetics
PASLI disease is caused by gain-of-function mutations in the gene PIK3CD, which stands for phosphatidylinositol 3-kinase, catalytic, delta.[3] PIK3CD maps to human chromosome 1p36.2 [4] and encodes the p110δ catalytic PI(3)K subunit. The p110δ subunit is a protein of 1,044 amino acids that is predominantly expressed in leukocytes and plays a role in adaptive immunity. PI(3)K enzymes are activated by a variety of cell surface receptors including antigen receptors on lymphocytes. Once activated, they phosphorylate inositol lipids in the cell membrane, which triggers additional downstream signaling events.
A variant of PASLI disease can all be caused by heterozygous splice site mutation in PIK3R1, which encodes the p85α, p55α, and p50α regulatory PI3K subunits. These patients suffer from recurrent sinopulmonary infections and lymphoproliferation, exhibit hyperactive PI3K signaling, and have prominent expansion and skewing of peripheral blood CD8+ T cells toward terminally differentiated, senescent effector cells with short telomeres.[5]
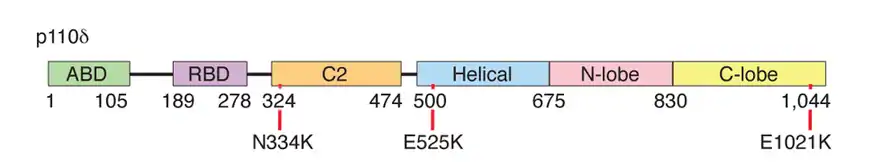
P110δ contains at least five domains (figure 1): adaptor-binding domain, a Ras GTPase-binding domain, a PI(3)K-type C2 domain, helical domain, and a kinase domain.[1] Mutations have been identified in multiple domains,[1] although there seems to be a recurrent transition mutation (G>A causing E1021K) in the C lobe of the kinase domain,[1][2] leading to constitutive activation of enzyme function. Specific p110δ mutants cause stronger binding to membranes and relieve inhibition of the kinase by regulatory proteins.[1][2] These changes appear to affect the immune system through over activating the downstream mTOR pathway.[1]
Inheritance
PASLI disease is inherited in an autosomal dominant manner. This means one only needs a single abnormal gene from his/her parents to have PASLI disease. Of the two copies of PIK3CD each person carries, the abnormal PIK3CD gene dominates despite the fact that the matching PIK3CD gene from the other parent is normal. Additionally, dominant inheritance means most families with PASLI disease have affected relatives in each generation on the side of the family with the mutation. An alternative type of PIK3CD mutation is called de novo, which means that the mutation was not inherited from the parents but rather spontaneously arose in the patient.
Children of a parent who carries a PIK3CD mutation have a 50% chance of inheriting the mutation. In a family, each child’s risk of inheriting the mutated PIK3CD gene is independent of whether or not other siblings have the mutation. For example, if the first three children in a family have the mutation, the next child has the same 50% risk of inheriting the mutation. Children who do not inherit the abnormal gene will not develop PASLI disease or pass on the mutation.
The variant of PASLI disease due to mutations in PIK3R1 is also inherited in an autosomal dominant manner.
Diagnosis
The clinical symptoms are caused by immunological abnormalities (figure 2). These include deficiency in CD27+ B memory cells, overrepresentation of CD10+ transitional B cells, expanded effector (CCR7-) T cells, expanded CD57+ senescent CD8+ T cells, and alterations in serum immunoglobulin concentrations, most with normal to elevated concentrations of IgM and reduced concentrations of IgA.[1]
Treatment
Once a diagnosis is made, the treatment is based on an individual’s clinical condition. Based on the apparent activation of the mTOR pathway, Lucas and colleagues [1] treated patients with rapamycin, an mTOR inhibitor. This effectively reduced hepatosplenomegaly and lymphadenopathy, most likely by restoring the normal balance of naïve, effector, and memory cells in the patients’ immune system. More research is needed to determine the most effective timing and dosage of this medication and to investigate other treatment options. Investigators at the National Institute of Allergy and Infectious Diseases at the US National Institutes of Health currently have clinical protocols to study new approaches to the diagnosis and treatment of this disorder.[6]
References
- Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, Avery DT, Moens L, Cannons JL, Biancalana M, Stoddard J, Ouyang W, Frucht DM, Rao VK, Atkinson TP, Agharahimi A, Hussey AA, Folio LR, Olivier KN, Fleisher TA, Pittaluga S, Holland SM, Cohen JI, Oliveira JB, Tangye SG, Schwartzberg PL, Lenardo MJ, Uzel G (2014). "Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency". Nature Immunology. 15 (1): 88–97. doi:10.1038/ni.2771. PMC 4209962. PMID 24165795.
- Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, Baxendale H, Coulter T, Curtis J, Wu C, Blake-Palmer K, Perisic O, Smyth D, Maes M, Fiddler C, Juss J, Cilliers D, Markeli G, Chandra A, Farmer G, Kielkowska A, Clark J, Kracker S, Debré M, Picard C, Pellier I, Jabado N, Morris JA, Barcenas-Morales G, Fischer A, Spehens L, Hawkins P, Barrett JC, Abinum M, Clatworthy M, Durandy A, Doffinger R, Chilvers ER, Cant AJ, Kumararatne D, Okkenhaug K, Williams RL, Condliffe A, Nejentsev S (2013). "Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage". Science. 342 (6160): 866–871. Bibcode:2013Sci...342..866A. doi:10.1126/science.1243292. PMC 3930011. PMID 24136356.
- "OMIM: PHOSPHATIDYLINOSITOL 3-KINASE, CATALYTIC, DELTA; PIK3CD". OMIM. Johns Hopkins University. Retrieved December 8, 2018.
- Seki N; Nimura Y; Ohira M; Saito T; Ichimiya S; Nomura N; Nakagawara A (1997). "Identification and chromosome assignment of a human gene encoding a novel phosphatidylinositol-3 kinase". DNA Res. 4 (5): 355–358. doi:10.1093/dnares/4.5.355. PMID 9455486.
- Lucas, Carrie L.; Zhang, Yu; Venida, Anthony; Wang, Ying; Hughes, Jason; McElwee, Joshua; Butrick, Morgan; Matthews, Helen; Price, Susan; Biancalana, Matthew; Wang, Xiaochuan; Richards, Michael; Pozos, Tamara; Barlan, Isil; Ozen, Ahmet; Rao, Koneti; Su, Helen; Lenardo, Michael (2014). "Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K". Journal of Experimental Medicine. 211 (13): 2537–2547. doi:10.1084/jem.20141759. PMC 4267241. PMID 25488983.
- Clinicaltrials.gov, study ID: NCT00246857 and NCT00001467
External links
| Classification |
|---|