Peptide-mass fingerprint
In bio-informatics, a peptide-mass fingerprint or peptide-mass map is a mass spectrum of a mixture of peptides that comes from a digested protein being analyzed. The mass spectrum serves as a fingerprint in the sense that it is a pattern that can serve to identify the protein.[1] The method for forming a peptide-mass fingerprint, developed in 1993, consists of isolating a protein, breaking it down into individual peptides, and determining the masses of the peptides through some form of mass spectrometry.[2] Once formed, a peptide-mass fingerprint can be used to search in databases for related protein or even genomic sequences, making it a powerful tool for annotation of protein-coding genes.[3]
One major advantage to mass fingerprinting is that it is significantly faster to carry out than peptide sequencing, yet the results are equally useful.[4] Disadvantages include the need for a single protein for analysis and the requirement that the protein sequence is located, at least with significant homology, in a database. Because the mass of individual peptides is measured in forming a fingerprint, mixtures of different proteins can yield unreliable results. Therefore, sample preparation is an important step in the process. Even then, if reliable results are obtained, there must be a matching peptide sequence in the database you are searching in order for the results to be useful.[5]
Sample preparation
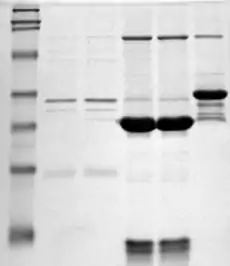
Before analyzing with mass spectrometry, a protein must be accurately isolated and digested. If not isolated, the results will represent a mixture of two or more proteins and will therefore be unreliable in protein identification. Because of this sensitivity, sample preparation is likely the most important step in forming a peptide-mass fingerprint.
Isolation of a specific protein is most often done through a form of gel electrophoresis, in which proteins are separated by size and can be subsequently extracted for further preparation. However, they can also be isolated by liquid chromatography. This method also separates proteins by size.[6]
Once an individual protein is isolated, it needs to be digested and fractionated for further analysis by a spectrometer. This is done by the addition of proteolytic enzymes such as trypsin and chymotrypsin.[7]
Another method commonly used that combines both the isolation and digestion steps is SDS-PAGE, a form of electrophoresis that separates and fractionates proteins simultaneously.
Spectrometric analysis
The digested protein can be analyzed with different types of mass spectrometers such as ESI-TOF or MALDI-TOF. MALDI-TOF is often the preferred instrument because it allows a high sample throughput and several proteins can be analyzed in a single experiment, if complemented by MS/MS analysis.[8]
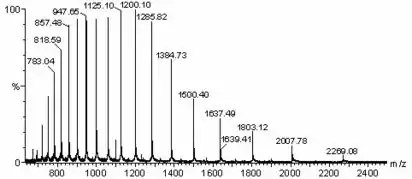
In matrix-assisted laser desorption ionization (MALDI), a fragmented peptide sample is loaded onto a matrix and ionized through the use of a high energy laser. The fragmented ions are then separated by mass-to-charge ratio based on the time of flight (TOF) through the spectrometer. They can then be further fragmented and re-analyzed in tandem mass spectrometry, often with a quadrupole ion trap,[9] but also possible with tandem time of flight.[10]
The output received from a mass spectrometer comes in the form of a peak list. This spectrum shows the masses and relative abundances of the peptide fragments present in the sample. In reading a spectrum like the one shown, all possible major fragmentations of a protein would need to be considered. Then the masses of those fragments would correlate to the numbers in the peaks of the spectrum. While it can be analyzed to some degree on its own, in forming a peptide-mass fingerprint, the peak list is run through a database search to find homologous peptide sequences.
Computer database analysis
The peak list obtained through spectrometric means is used as the query in a database search using the software MASCOT.[11] The MASCOT software uses an algorithm that looks for significant peptide sequence homology to present the most statistically likely protein in the sample, based on the results.
In performing the search, you much choose a database to go through. Such databases include, among others, Swissprot, often used when researching well characterized organisms like humans, mice, and yeasts; and NCBInr for more general, robust searches.
A detailed tutorial on using MASCOT software can be found in a link below.
Applications
The use of a peptide-mass fingerprint is fairly widespread in proteomic research. Some specific examples of how it has been used in the field are as follows:
Screening and characterization of amylase and cellulase activities in psychrotolerant yeasts
The authors of this study sought to determine which yeasts were metabolically active at lower temperatures and could therefore be used for colder industrial processes. They grew various yeasts on medium at different temperatures, then determined enzyme activity by separating proteins on a gel and fingerprinting the individual bands. Through database search they found the enzyme of interest and discovered two individual yeasts that had higher activity at lower temperatures.[12]
APOA-I: A Possible Novel Biomarker for Metabolic Side Effects in First Episode Schizophrenia
The authors of this study sought to determine the effect on metabolism of the drug risperidone in schizophrenia patients. After discovering that risperidone did have negative metabolic side effects, they tested membrane proteins for glucose and lipid transport in control and experimental groups by MALDI-TOF and fingerprinting. Results showed altered fingerprints and therefore altered levels of folding in the proteins. So, they concluded that risperidone negatively effects glucose and lipid transport proteins in the cell membranes of patients.[13]
References
- Mass spectrometry in the biological sciences
- James, P.; Quadroni, M.; Carafoli, E.; Gonnet, G. (1993-08-31). "Protein identification by mass profile fingerprinting". Biochemical and Biophysical Research Communications. 195 (1): 58–64. doi:10.1006/bbrc.1993.2009. ISSN 0006-291X. PMID 8363627.
- Cottrell, J. S. (1994-06-01). "Protein identification by peptide mass fingerprinting". Peptide Research. 7 (3): 115–124. ISSN 1040-5704. PMID 8081066.
- Pappin, D. J.; Hojrup, P.; Bleasby, A. J. (1993-06-01). "Rapid identification of proteins by peptide-mass fingerprinting". Current Biology. 3 (6): 327–332. doi:10.1016/0960-9822(93)90195-t. ISSN 0960-9822. PMID 15335725. S2CID 40203243.
- Henzel, William J; Watanabe, Colin; Stults, John T (2003-09-01). "Protein identification: the origins of peptide mass fingerprinting". Journal of the American Society for Mass Spectrometry. 14 (9): 931–942. doi:10.1016/S1044-0305(03)00214-9. PMID 12954162.
- "EMBnet node Switzerland" (PDF). www.ch.embnet.org. Retrieved 2016-04-11.
- Yates, J. R.; Speicher, S.; Griffin, P. R.; Hunkapiller, T. (1993-11-01). "Peptide mass maps: a highly informative approach to protein identification". Analytical Biochemistry. 214 (2): 397–408. doi:10.1006/abio.1993.1514. ISSN 0003-2697. PMID 8109726.
- Yates, J. R. (1998-01-01). "Mass spectrometry and the age of the proteome". Journal of Mass Spectrometry. 33 (1): 1–19. Bibcode:1998JMSp...33....1Y. doi:10.1002/(SICI)1096-9888(199801)33:1<1::AID-JMS624>3.0.CO;2-9. ISSN 1076-5174. PMID 9449829.
- Ahmed, Farid E. (2008-12-01). "Utility of mass spectrometry for proteome analysis: part I. Conceptual and experimental approaches". Expert Review of Proteomics. 5 (6): 841–864. doi:10.1586/14789450.5.6.841. ISSN 1744-8387. PMID 19086863. S2CID 38574652.
- Vestal, Marvin L.; Campbell, Jennifer M. (2005-01-01). Tandem time-of-flight mass spectrometry. Methods in Enzymology. 402. pp. 79–108. doi:10.1016/S0076-6879(05)02003-3. ISBN 9780121828073. ISSN 0076-6879. PMID 16401507.
- Wright, James C.; Collins, Mark O.; Yu, Lu; Käll, Lukas; Brosch, Markus; Choudhary, Jyoti S. (2012-08-01). "Enhanced peptide identification by electron transfer dissociation using an improved Mascot Percolator". Molecular & Cellular Proteomics. 11 (8): 478–491. doi:10.1074/mcp.O111.014522. ISSN 1535-9484. PMC 3412976. PMID 22493177.
- Carrasco, Mario; Villarreal, Pablo; Barahona, Salvador; Alcaíno, Jennifer; Cifuentes, Víctor; Baeza, Marcelo (2016-02-19). "Screening and characterization of amylase and cellulase activities in psychrotolerant yeasts". BMC Microbiology. 16: 21. doi:10.1186/s12866-016-0640-8. ISSN 1471-2180. PMC 4759947. PMID 26895625.
- Song, Xueqin; Li, Xue; Gao, Jinsong; Zhao, Jingping; Li, Youhui; Fan, Xiaoduo; Lv, Luxian (2014-04-07). "APOA-I: A Possible Novel Biomarker for Metabolic Side Effects in First Episode Schizophrenia". PLOS ONE. 9 (4): e93902. Bibcode:2014PLoSO...993902S. doi:10.1371/journal.pone.0093902. ISSN 1932-6203. PMC 3978061. PMID 24710015.
External links
- https://www.bruker.com/fileadmin/user_upload/8-PDF-Docs/Separations_MassSpectrometry/InstructionForUse/8702557_IFU_Bruker_Guide_MALDI_Sample_Preparation_Revision_E.pdf
- http://www.matrixscience.com/help/pmf_help.html
- http://www.matrixscience.com/cgi/search_form.pl?FORMVER=2&SEARCH=PMF
- https://www.youtube.com/watch?v=xh8GGzsc2r4