Terminal amine isotopic labeling of substrates
Terminal amine isotopic labeling of substrates (TAILS) is a method in quantitative proteomics that identifies the protein content of samples based on N-terminal fragments of each protein (N-terminal peptides) and detects differences in protein abundance among samples.
Like other methods based on N-terminal peptides, this assay uses trypsin to break proteins into fragments and separates the N-terminal peptides (the fragments containing the N-termini of the original proteins) from the other fragments (internal tryptic peptides). TAILS isolates the N-terminal peptides by identifying and removing the internal tryptic peptides. This negative selection allows the TAILS method to detect all N-termini in the given samples. Alternative methods that rely on the free amino group of the N-terminus to identify the N-terminal peptides cannot detect some N-termini because they are "naturally blocked" (i.e. the natural protein does not have a free amino group).
The TAILS method has a number of applications including the identification of new substrates and proteases (including those that have an unknown and broad specificity)[1] and as a way to define the termini of proteins that enables protein annotation. TAILS can also be used to link proteases with a variety of defined biological pathways in diseases such as cancer, in order to gain a clearer understanding of the substrates and proteases involved in the disease state.[2]
Method
TAILS is a 2D or 3D proteomics based assay for the labeling and isolation of N-terminal peptides, developed by a group at the University of British Columbia.[1] The TAILS method is designed for comparison of multiple protease treated cells and control proteome cells.[2] Samples can be derived from a variety of sources including tissue, fibroblasts, cancer cells and from fluid effusions.
This assay isolates the N-terminal peptides by removing the internal tryptic peptides via ultrafiltration leaving the labeled mature N-terminal and neo-N-Terminal peptides to be analyzed by tandem mass spectrometry (MS/MS). This negative selection allows the TAILS method to detect all N-termini in the given samples. Alternative methods that rely on the free amino group of the N-terminus to isolate the N-terminal peptides cannot detect naturally blocked N-termini because they do not have a free amino group.
TAILS requires only small sample of peptide for experimentation (100–300 ug), can be used with proteases which have unknown or broad specificity and supports a variety of methods for sample labeling. However, it identifies ~ 50% of proteins by two or more different and unique peptides (one of the original mature N-terminus and/or one or more neo-N-terminal peptide via cleavage site) that do not represent independent biological events thus cannot be averaged for quantification. It also has difficulty validating results for single peptide-base N-terminome analysis.[1]
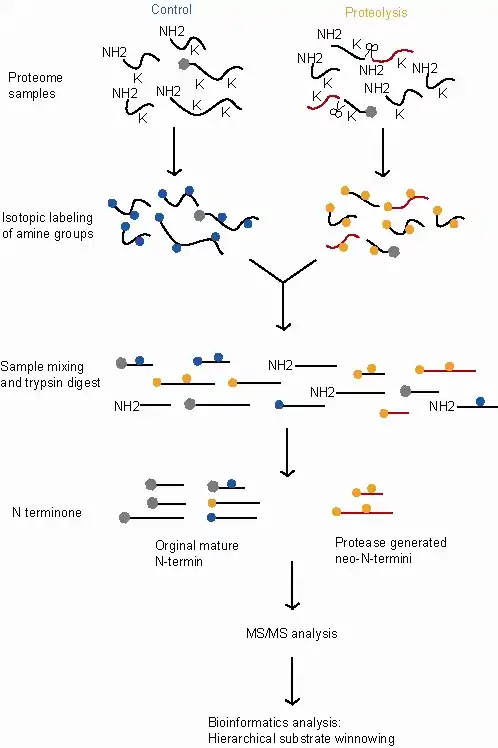
The following steps are for the dimethylation-TAILS assay, comparing a control sample (exhibiting normal proteolytic activity) and a treated sample (which in this example exhibits an additional proteolytic activity).
- Proteome-wide proteolysis occurring in both the treated and control samples with additional proteolytic activity in the treated sample.
- Inactivation of the proteases and protein denaturation and reduction.
- Labelling with stable isotopes. This allows peptides that originated in the control sample to be distinguished from those that originated in the treated sample so their relative abundance can be compared. In this example, the labelling is applied by reductive dimethylation of the primary amines using either heavy (d(2)C13)-formaldehyde for the treated samples or light (d(O)C12)-formaldehyde for the controls. This reaction is catalyzed by sodium cyanoborohydride and attaches the labeled methyl groups to lysine-amines and the free (∝)- amino groups at the N-termini of the proteins and protease cleavage products.
- Blocking of reactive amino groups. This allows the internal tryptic peptides to be identified later in the process because they will be the only peptides with reactive amino groups. In this example the labelling reaction (reductive dimethylation) also blocks the reactive amino groups.
- Pooling. The two labeled proteomes are now mixed. This ensures that the samples are treated identically at all subsequent steps allowing the relative quantities of the proteins in the two samples to be more accurately measured.
- Trypsinization. This breaks each protein into fragments. The labeled N-termini of the original proteins remain blocked, while the new internal tryptic peptides have a free N-terminus.
- Negative selection. A hyperbranched polyglycerol and aldehyde (HPG) polymer specific for tryptic peptide binding is added to the sample and reacts with the newly generated tryptic peptides through their free N-termini. As in step 3 above, this reaction is catalyzed by sodium cyanoborohydride. The dimethylated lysine's acetylated and isotopically labeled protein peptides and neo(new)-N-terminal peptides are unreactive and remain unbound and can be separated from the poly-internal tryptic peptide complexes using ultrafiltration.
- The eluted unbound proteins are highly concentrated with the N-terminal peptides and neo-N-terminal peptides.
- This eluted sample is then quantified and analysis completed by MS/MS.
- The final step in TAILS involves bioinformatics. Using a hierarchical substrate winnowing process that discriminates from background proteolysis products and non-cleaved proteins by a peptide isotope quantification and certain bioinformatic search criteria.[1][2]
Types
The types of TAILS differ in the methods used to block and label the amino groups of the proteins and protease cleavage products. These amino groups include lysine-amines and the free (∝)-amino groups of the N-termini of the proteins.
Dimethylation-TAILS procedure is a chemical labeling based procedure that is performed in one step using amine-reactive isotopic reagents. The labeling of two samples uses either 12CH2-formaldehyde (light) or 13CD2-formaldehyde (heavy) and uses sodium cyanoborohydride as the catalyst.[1] The advantage of this method is that it is robust, efficient and cost effective.The labeling procedure for the controls and protease treated samples must be carried out separately before they can be pooled, and it is limited to two samples per experiment, which may be a disadvantage if multiple samples need to be studied simultaneously.[1]
Stable isotope labeling with amino acids in cell culture (SILAC) is a procedure that can be done in vivo. This procedure can be used in all cell culture laboratories and is a routinely used labeling technique. This metabolic labeling enables inhibition of a given protease in biological samples and analysis of ex vivo processing.[1] An advantage of using this metabolic labeling method over chemical labeling is that it allows for reliable, fast and efficient discrimination between the real cellular derived proteins that are being investigated from contaminants such as serum proteins. SILAC TAILS can be used for analysis of up to five multiplex samples. SILAC is not suitable for clinically relevant human samples that are not able to be metabolically labeled. SILAC is an expensive method and may not be a feasible option for most laboratories.[1]
The isobaric tag for relative and absolute quantification (iTRAQ) method or iTRAQ-TAILS enables the quantitaion of multiple samples simultaneously.This method has the ability to simultaneously analyze from 4-8 samples in multiplex experiments using four- and eight- plex iTRAQ reagents.This method provides high accuracy identification and quantification of samples and allows for more reproducible analysis of sample replicates.[1] Like other iTRAQ methods, iTRAQ-TAILS requires a MALDI mass spectrometer and costly iTRAQ reagents.
Alternative methods
There are several alternative approaches to studying N termini and proteolysis products.
Acetylation of amines followed by tryptic digestion and biotinylation of free N-terminal peptides uses chemical (acetylation) to label free lysines and N-termini. The blocked N-termini is then negatively selected. However, the naturally free internal N-termini and blocked N-termini cannot be distinguished after acetylation. This method does not use isotopic labeling, thus it is difficult to quantify the findings. Also, it is hard distinguish between experimental and background proteolysis products.[3]
Lysine guanidination followed by biotinylation of N-termini uses a chemical to block lysine residues and tag free N-termini. The tagged free N-termini is then selected. The down side to this method is that the findings cannot be applied to a statistical model using non-cleaved peptides due to not being able to capture naturally blocked N-termini. Since it does not involve isotopic labeling, the results cannot be quantified. The cleavage site also has to be already known to do labeling.[4]
Subtiligase biotinylation of N-termini uses enzymatic labeling of N-terminal peptides, but does not use lysine blocking chemicals. Without lysine blocking, many of the cleaved N-terminal peptide will be too short for identification. The results can be highly dependent on the properties of subtiligase thus may be biased. This method does not capture naturally blocked N-termini and also does not use isotopic labeling thus it would be difficult to quantify findings.[5]
ITRAQ-labeling of N-termini uses iTRAQ to label the N-termini. Neo-N-termini peptides are selected through in silico. The down side to this technique is that MALDI mass spectrometer is needed and the iTRAQ reagents required are costly. This method does not capture naturally blocked N-termini. The whole process will require 50-100mg of peptide samples.[6]
Combined fractional diagonal chromatography (COFRADIC) allows different labeling for naturally blocked N-termini and protease generated neo-N-termini. All blocked N-termini are negatively selected. However the process requires many chemical processing, chromatography and mass spectrometry. The best separation results are dependent on the amino acid modification such as methionine oxidation not occurring during handling. This method requires 150 MS/MS analyses per sample but samples can be pooled for mass spectrometry (and number of analyses can be reduced). This technique is suitable to be used for proteases with unknown or broad specificity.[7]
References
- Kleifeld, Oded; Doucet, Alain; Prudova, Anna; Auf Dem Keller, Ulrich; Gioia, Magda; Kizhakkedathu, Jayachandran N; Overall, Christopher M (2011). "Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates". Nature Protocols. 6 (10): 1578–611. doi:10.1038/nprot.2011.382. PMID 21959240.
- Kleifeld, Oded; Doucet, Alain; Auf Dem Keller, Ulrich; Prudova, Anna; Schilling, Oliver; Kainthan, Rajesh K; Starr, Amanda E; Foster, Leonard J; et al. (2010). "Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products". Nature Biotechnology. 28 (3): 281–8. doi:10.1038/nbt.1611. PMID 20208520.
- McDonald, Lucy; Robertson, Duncan H L; Hurst, Jane L; Beynon, Robert J (2005). "Positional proteomics: Selective recovery and analysis of N-terminal proteolytic peptides". Nature Methods. 2 (12): 955–7. doi:10.1038/nmeth811. PMID 16299481.
- Timmer, John C.; Enoksson, Mari; Wildfang, Eric; Zhu, Wenhong; Igarashi, Yoshinobu; Denault, Jean-Benard; Ma, Yuliang; Dummitt, Benjamin; et al. (2007). "Profiling constitutive proteolytic events in vivo". Biochemical Journal. 407 (1): 41–8. doi:10.1042/BJ20070775. PMC 2267409. PMID 17650073.
- Mahrus, Sami; Trinidad, Jonathan C.; Barkan, David T.; Sali, Andrej; Burlingame, Alma L.; Wells, James A. (2008). "Global Sequencing of Proteolytic Cleavage Sites in Apoptosis by Specific Labeling of Protein N Termini". Cell. 134 (5): 866–76. doi:10.1016/j.cell.2008.08.012. PMC 2566540. PMID 18722006.
- Enoksson, Mari; Li, Jingwei; Ivancic, Melanie M.; Timmer, John C.; Wildfang, Eric; Eroshkin, Alexey; Salvesen, Guy S.; Tao, W. Andy (2007). "Identification of Proteolytic Cleavage Sites by Quantitative Proteomics". Journal of Proteome Research. 6 (7): 2850–8. doi:10.1021/pr0701052. PMID 17547438.
- Gevaert, Kris; Goethals, Marc; Martens, Lennart; Van Damme, Jozef; Staes, An; Thomas, Grégoire R.; Vandekerckhove, Joël (2003). "Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides". Nature Biotechnology. 21 (5): 566–9. doi:10.1038/nbt810. PMID 12665801.