Upshaw–Schulman syndrome
Upshaw–Schulman syndrome (USS) is the recessively inherited form of thrombotic thrombocytopenic purpura (TTP), a rare and complex blood coagulation disease. USS is caused by the absence of the ADAMTS13 protease resulting in the persistence of ultra large von Willebrand factor multimers (ULVWF), causing episodes of acute thrombotic microangiopathy with disseminated multiple small vessel obstructions.[1][2] These obstructions deprive downstream tissues from blood and oxygen, which can result in tissue damage and death. The presentation of an acute USS episode is variable but usually associated with thrombocytopenia, microangiopathic hemolytic anemia (MAHA) with schistocytes on the peripheral blood smear,[3] fever and signs of ischemic organ damage in the brain, kidney and heart.
| Upshaw–Schulman syndrome | |
|---|---|
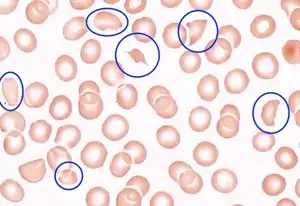 | |
| Blood smear under the microscope with typical schistocytes in TTP marked in blue – H&E stain | |
| Specialty | Rheumatology |
Signs and symptoms
The presentation of TTP is variable. The initial symptoms, which force the patient to medical care, are often the consequence of lower platelet counts like purpura (present in 90% of patients), ecchymosis and hematoma.[4] Patients may also report signs and symptoms as a result of (microangiopathic) hemolytic anemia, such as (dark) beer-brown urine, (mild) jaundice, fatigue and pallor. Cerebral symptoms of various degree are present in many patients, including headache, paresis, speech disorder, visual problems, seizures and disturbance of consciousness up to coma. The symptoms can fluctuate so that they may only be temporarily present but may reappear again later in the TTP episode. Other unspecific symptoms are general malaise, abdominal, joint and muscle pain. Severe manifestations of heart or lung involvements are rare, although affections are not seldom measurable (such as ECG-changes).[5]
Cause
Genetics
The ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif 13) gene is located on chromosome 9q34 and encoding 29 exons.[6] The ADAMTS13 protease consists of 1427 amino acids and has several protein domains:[1][6]
- The signal peptide is thought to have a role in the secretion, folding and stability of the ADAMTS13 protein.[7] It interacts with membrane phospholipids and protein components of the secretory machinery within the cells.[7]
- The metalloprotease domain contains the active site, which cleaves the unfolded VWF within the A2 domain, between the amino acids Tyrosine1605 and Methionine1606.[8]
- The disintegrin domain together with the TSP1 repeat, the following cysteine-rich and spacer domains are necessary for substrate recognition, binding and cleavage[7] of the VWF-A2 domain.[8] First, the spacer domain recognizes VWF, increasing the affinity of ADAMTS13 to VWF. Next the disintegrin-like domain engages in a low-affinity binding. Then the metalloprotease domain interacts with the VWF, similar to a three-step molecular zipper.[9]
- The TSP1 repeats mediate extracellular matrix protein-protein interactions.[5]
- The cystein-rich domain is responsible for attachment, for example, to integrins of different cell membranes.[5]
- The CUB domains take part in protein-protein interactions[5] with VWF domains, which become exposed under shear stress,[8] and are also involved in the binding and cleavage of VWF.[7] Additionally they are involved in the ADAMTS13 protein secretion[10]
Disease causing mutations in ADAMTS13, which can be found in all ADAMTS13 protease domains.[8] result predominantly in impaired ADAMTS13 secretion with or without decreased ADAMTS13 protease activity.[7] More than 120 disease causing mutations and numerous single-nucleotide polymorphisms (SNP) are known today.[11][12] Residual ADAMTS13 activity has been observed with certain mutations and seems to be associated with a later disease-onset.[11] It has been postulated that some SNPs interact with each other and may amplify or reduce overall ADAMTS13 activity.[6][7]
ADAMTS13 function and pathogenesis
The ADAMTS protease family contains enzymes that process collagen, cleave inter-cellular matrix, inhibit angiogenesis and blood coagulation. ADAMTS13 belongs to the zinc metalloproteases, and is mainly expressed in liver stellate cells and endothelial cells, but was also found in other cell types, such as platelets, podocytes in the kidney and several brain cells.[8][13][14] The only known role of the ADAMTS13 protease is to cleave VWF multimers.[15] The plasma half-life of administered ADAMTS13 in USS patients is around 2–4 days, whereas the protective effects seems to last longer.[16][17]
Usually USS patients have a severely deficient ADAMTS13 activity of <10% of the normal.[9] In this low range there may be residual ADAMTS13 activity, depending on the underlying mutations.[6][7][11]
In USS severe ADAMTS13 deficiency is often not enough to induce a (first) acute TTP episode. It primarily occurs when an additional (environmental) trigger is present.[1][6][8] Recognized triggers are infections (including mild flu-like upper airway infections), pregnancy,[18] heavy alcohol intake[16] or certain drugs.[1][9] In these situations, VWF is released from its storage organelles, such as Weibel–Palade bodies and granules of platelets. Increased VWF levels in the circulation are leading to a higher demand of ADAMTS13, which is lacking in USS, and can bring forward a TTP episode.[3][5]
Pathology
After secretion, ADAMTS13 is either bound to the endothelial surface or free in the blood stream.[3] The heightened shear stress in small- and microvessels alters the 3D-structure of VWF from the contracted globular form to its linear form.[8] The linear VWF has now its active binding sites exposed, that are important to start blood coagulation.[8] These sites bind platelets and blood vessel lesions by interlinking the stretched VWF with one another – a blood clot is formed.[8] In its uncut form, (ultra large) VWF's heightened stickiness and interlinking causes spontaneous platelet binding and blood clotting.[8] The linear VWF exposes the A2 domain, so that in the presence of enough ADAMTS13 activity it gets cut to its normal size.[3] VWF in the normal length loses its heightened stickiness and spontaneous crosslinking activity to only form blood clots when needed.[3]
Diagnosis
A diagnosis of TTP is based on the clinical symptoms with the concomitant presence of thrombocytopenia (platelet count below 100×109/L) and microangiopathic hemolytic anemia with schistocytes on the blood smear, a negative direct antiglobulin test (Coombs test), elevated levels of hemolysis markers (such as total bilirubin, LDH, free hemoglobin, and an unmeasurable haptoglobin), after exclusion of any other apparent cause.[1][2]
USS can present similar to the following diseases, which have to be excluded: fulminant infections, disseminated intravascular coagulation, autoimmune hemolytic anemia, Evans syndrome, the typical and atypical form of hemolytic uremic syndrome, HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome, pre-eclampsia, heparin-induced thrombocytopenia, cancer that is often accompanied with metastasis, kidney injury, antiphospholipid antibody syndrome, and side effects from hematopoietic stem cell transplantation.[1][2]
Of note is that pregnancy associated affections like pre-eclampsia, eclampsia, and HELLP syndrome can overlap in their presentation as pregnancy can trigger TTP episodes.[19]
Patients with fulminant infections, disseminated intravascular coagulation, HELLP syndrome, pancreatitis, liver disease, and other active inflammatory conditions may have reduced ADAMTS13 activity, but almost never a relevant severe ADAMTS13 deficiency <10% of the normal.[2][20]
A severe ADAMTS13 deficiency below 5% or <10% of the normal (depending on the definitions)[1][21] is highly specific for the diagnosis of TTP.[16][22] ADAMTS13 activity assays are based on the direct or indirect measurement of VWF-cleavage products. Its activity should be measured in blood samples taken before therapy has started, to prevent false high ADAMTS13 activity.[2] If a severe ADAMTS13 deficiency is present an ADAMTS13 inhibitor assay is needed to distinguish between the acquired, autoantibody-mediated and the congenital form of TTP (USS).[1] The presence of antibodies can be tested by ELISA or functional inhibitor assays. The level of ADAMTS13 inhibitor may be fluctuating over the course of disease and depends on free circulatory antibodies, therefore a onetime negative test result does not always exclude the presence of ADAMTS13 inhibitors and thereby an autoimmune origin of TTP.[2] A severe ADAMTS13 deficiency in the absence of an inhibitor, confirmed on a second time point in a healthy episode of a possible USS patient, usually sets the trigger to perform a molecular analysis of the ADAMTS13 gene to confirm a mutation. In unclear cases a plasma infusion trial can be done, showing an USS in the absence of anti-ADAMTS13-antibodies a full recovery of infused plasma-ADAMTS13 activity as well as a plasma half-life of infused ADAMTS13 activity of 2–4 days. A deficiency of ADAMTS13 activity in first-degree relatives is also a very strong indicator for an Upshaw-Schulman syndrome.[1][2][9]
Treatment
The therapy of an acute TTP episode has to be started as early as possible.[1][2] The standard treatment is the daily replacement of the missing ADAMTS13 protease in form of plasma infusions or in more severe episodes by plasma exchange. In the latter the patients plasma is replaced by donated plasma.[23] The most common sources of ADAMTS13 is platelet-poor fresh frozen plasma (FFP)[3] or solvent-detergent plasma.
The benefit of plasma exchange compared to plasma infusions alone may result from the additional removal of ULVWF.[1][23] In general both plasma therapies are well tolerated, several mostly minor complications may be observed.[2][23] The number of infusion/exchange sessions needed to overcome a TTP episode are variable but usually take less than a week in USS.[1][9] The intensive plasma-therapy is generally stopped when platelet count increases to normal levels and is stable over several days.[1][2]
Preventive therapy
Not all affected patients seem to need a regular preventive plasma infusion therapy, especially as some reach long-term remission without it.[24] Regular plasma infusions are necessary in patients with frequent relapses and in general situations with increased risk to develop an acute episode (as seen above) such as pregnancy. Plasma infusions are given usually every 2-3 weeks to prevent acute episodes of USS,[16] but are often individually adapted.
Outlook
Several therapy developments for TTP emerged during recent years. Artificially produced ADAMTS13 has been used in mice[25] and testing in humans has been announced. Another drug in development is targeting VWF and its binding sites, thereby reducing VWF-platelet interaction, especially on ULVWF during a TTP episode.[26] Among several (multi-)national data bases a worldwide project has been launched to diagnose USS patients and collect information about them to gain new insights into this rare disease with the goal to optimize patient care.[27]
Epidemiology
The incidence of acute TTP in adults is around 1.7–4.5 per million and year.[28][29] These cases are nearly all due to the autoimmune form of TTP, where autoantibodies inhibit ADAMTS13 activity.[1][9] The prevalence of USS has not yet been determined, but is assumed to constitute less than 5% of all acute TTP cases. The syndrome's inheritance is autosomal recessive, and is more often caused by compound heterozygous than homozygous mutations.[30] The age of onset is variable and can be from neonatal age up to the 5th–6th decade.[1] The risk of relapses differs between affected individuals.[1][9] Minimization of the burden of disease can be reached by early diagnosis and initiation of prophylaxis if required.[2]
History

TTP was first recognized as a disease in 1947 and the name was given according to symptoms and underlying pathophysiology which differed from the already known immune thrombocytopenia.[16] Schulman reported a case of TTP in 1960[1] and Upshaw published a paper in 1978 about relapsing TTP in a patient whom he had followed for 11 years.[31] In his report Upshaw noted the similarities with the reported case by Schulman and hypothesized that the two cases had similar causes — a missing plasma factor.[1][31]
One year later the disease was named Upshaw-Schulmann Syndrome.[32] In 1996 the VWF-cleaving protease was discovered and in the following year found to be the major issue in TTP's pathogenesis.[15][33][34] In 2001 the VWF-cleaving protease was identified as ADAMTS13, the gene was mapped to chromosome 9q34, and the first USS-causing mutations were identified.[1][17]
References
- Sadler, J. E. (2008). "Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura". Blood. American Society of Hematology. 112 (112): 11–18. doi:10.1182/blood-2008-02-078170. PMC 2435681. PMID 18574040.
- Sarode, R.; Bandarenko, N.; Brecher, M.E.; Kiss, J.E.; Marques, M.B.; Szczepiorkowski, Z.M.; Winters, J.L. (June 2014). "Thrombotic thrombocytopenic purpura". Journal of Clinical Apheresis. 29 (3): 148–167. doi:10.1002/jca.21302. PMID 24136342.
- Moake, J.L. (August 22, 2002). "Mechanisms of Disease: Thrombotic Microangiopathies". The New England Journal of Medicine. 347 (8): 589–600. doi:10.1056/nejmra020528. PMID 12192020.
- George, J.N.; Chen, Q.; Deford, C.C.; Al-Nouri, Z. (2012). "Ten Patient Stories Illustrating the Extraordinarily Diverse Clinical Features of Patients With Thrombotic Thrombocytopenic Purpura and Severe ADAMTS13 Deficiency". Journal of Clinical Apheresis. 27 (6): 302–311. doi:10.1002/jca.21248. PMID 22927184.
- Galbusera, M.; Noris, M.; Remuzzi, G. (2006). "Thrombotoic thrombocytopenic Purpura – Then and Now". Seminars in Thrombosis and Haemostasis. 32 (2): 81–89. doi:10.1055/s-2006-939763. PMID 16575682.
- Camilleri, R.S.; Cohen, H.; Mackie, I.J.; Scully, M.; Starke, R.D.; Crawley, J.T.B.; Lane, D.A.; Machin, S.J. (2008). "Prevalence of the ADAMTS-13 missense mutation R1060W in late onset adult thrombotic thrombocytopenic purpura". Thrombosis and Haemostasis. 6 (2): 331–338. doi:10.1111/j.1538-7836.2008.02846.x. PMID 18031293.
- Plaimauer, B.; Fuhrmann, J.; Mohr, G.; Wernhart, W.; Bruno, K.; Ferrari, S.; Konetschny, Ch.; Antoine, G.; Rieger, M.; Scheiflinger, F. (January 1, 2006). "Modulation of ADAMTS13 secretion and specific activity by a combination of common amino acid polymorphisms and missense mutation". Blood. 107 (1): 118–124. doi:10.1182/blood-2005-06-2482. PMID 16160007.
- Lancellotti, S.; Bassao, M.; De Cristofaro, R. (September 2, 2013). "Proteolytic Processing of Von Willebrand Factor by ADAMTS13 and Leukocyte Proteases". Mediterranean Journal of Hematology and Infectious Diseases. 5 (1): e2013058. doi:10.4084/MJHID.2013.058. ISSN 2035-3006. PMC 3787661. PMID 24106608.
- Crawley, J.T.B.; Scully, M.A. (2013). "Thrombotic thrombocytopenic purpura: basic pathophysiology and therapeutic strategies". Hematology. American Society of Hematology. Education Program. American Society of Hematology. 2013: 292–299. doi:10.1182/asheducation-2013.1.292. PMID 24319194.
- Zhou, Z.; Xeh, H.; Jing, H.; Wang, C.; Tao, Z.; Choi, H.; Aboulfatova, K.; Li, R.; Dong, J. (2011). "Cystein residues in CUB-1 domain are critical for ADAMTS13 secretion and stability". Thrombosis and Haemostasis. 105 (1): 21–30. doi:10.1160/th10-07-0446. PMC 5649353. PMID 20886194.
- Lotta, LA; Wu, HM; Mackie, IJ; Noris, M; Veyradier, A; Scully, MA; Remuzzi, G; Coppo, P; Liesner, R; Donadelli, R; Loirat, C; Gibbs, RA; Horne, A; Yang, S; Garagiola, I; Musallam, KM; Peyvandi, F (July 12, 2012). "Residual plasmatic activity of ADAMTS13 is correlated with phenotype severity in congenital thrombotic thrombocytopenic purpura". Blood. 120 (2): 440–8. doi:10.1182/blood-2012-01-403113. PMC 3721603. PMID 22529288.
- Lotta, LA; Garagiola, I.; Palla, R.; Cairo, A.; Peyvandi, F. (January 2010). "ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura". Hum Mutat. 31 (1): 11–19. doi:10.1002/humu.21143. PMID 2010.
- Manea, M.; Kristoffersson, A.; Schneppenheim, R.; Saleem, M.A.; Mathieson, P.W.; Mörgelin, M.; Björk, P.; Holmberg, L.; Karpman, D. (September 2007). "Podocytes express ADAMTS13 in normal renal cortex and in patients with thrombotic thrombocytopenic purpura". British Journal of Haematology. 138 (5): 651–662. doi:10.1111/j.1365-2141.2007.06694.x. PMID 17627784.
- Tauchi, R.; Imagama, S.; Ohgomori, T.; Natori, T.; Shinjo, R.; Ishiguro, N.; Kadomatsu, K. (March 2012). "ADAMTS-13 is produced by glial cells and upregulated after spinal cord injury". Elvesier Neuroscience Letters. 517 (1): 1–6. doi:10.1016/j.neulet.2012.03.002. PMID 22425718. S2CID 5742973.
- Furlan, M.; Robles, R.; Lämmle, B. (May 15, 1996). "Partial Purification and Characterization of a Protease From Human Plasma Cleaving von Willebrand Factor to Fragments Produced by In Vivo Proteolysis". Blood. 87 (10): 4223–4234. doi:10.1182/blood.V87.10.4223.bloodjournal87104223. PMID 8639781.
- Kremer Hovinga, J.A.; Lämmle, B. (2012). "Role of ADAMTS13 in the pathogenesis, diagnosis and treatment of thrombotic thrombocytopenic purpura". Hematology. 2012 (1): 610–616. doi:10.1182/asheducation.V2012.1.610.3798654. PMID 23233642.
- Furlan, M.; Robles, R.; Morselli, B.; Sandoz, P.; Lämmle, B. (1999). "Recovery and Half-Life of von Willebrand Factor-Cleaving Protease after Plasma Therapy in Patients with Thrombotic Thrombocytopenic Purpura". Schattauer Verlag. 81 (1): 8–13. PMID 10348715.
- Jiang, Y.; Mclntosh, J.J.; Reese, J.A.; Deford, C.C.; Kremer Hovinga, J.A.; Lämmle, B.; Terrel, D.R.; Vessely, S.K.; Knudtson, E.J.; George, J.N. (March 13, 2014). "Pregnancy Outcomes Following Recovery Form Acquired Thrombotic Thrombocytopenic Purpura". Blood. American Society of Hematology. l123 (11): 1674–1680. doi:10.1182/blood-2013-11-538900. PMC 3954049. PMID 24398329.
- Yamashita, E.; Okada, H.; Yorioka, H.; Fujita, S.; Nishi, K.; Komiyama, Y.; Kanzaki, H. (March 2012). "Successful management of pregnancy-associated thrombotic thrombocytopenic purpura by monitoring ADAMTS13 activity". Journal of Obstetrics and Gynaecology Research. 38 (3): 567–569. doi:10.1111/j.1447-0756.2011.01742.x. PMID 22381108.
- Uemura, M.; Fujimura, Y.; Ko, S.; Matsumoto, M.; Nakajima, Y.; Fukui, H. (February 25, 2011). "Determination of ADAMTS13 and Its Clinical Significance for ADAMTS13 Supplementation Therapy to Improve the Survival of Patients with Decompensated Liver Cirrhosis". International Journal of Hepatology. 2011: 759047. doi:10.4061/2011/759047. PMC 3170842. PMID 21994870.
- George, JN; Al-Nouri, ZL (2012). "Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes". Hematology. 2012: 604–609. doi:10.1182/asheducation.V2012.1.604.3798564. PMID 23233641.
- Tripodi, A.; Peyvandi, F.; Chantarangkul, V.; Palla, R.; Afrasiabi, A.; Canciani, M.T.; Chung, D.W.; Ferrari, S.; Fujimura, Y.; Karimi, M.; Kokame, K.; Kremer Hovinga, J.A.; Lämmle, B.; De Meyer, S.F.; Plaimauer, B.; Vanhoorelbeke, K.; Varadi, K.; Mannucci, P.M. (2008). "Second international collaborative study evaluating performance characteristics of methods measuring the von Willebrand factor cleaving protease (ADAMTS-13)". Journal of Thrombosis and Haemostasis. 60 (9): 1676–1682. doi:10.1111/j.1538-7836.2008.03099.x. PMC 6537877. PMID 18662260.
- Rock, G.A.; Shumak, K.H.; Buskard, N.A.; Blanchette, Victor S.; Kelton, J.G.; Nair, R.C.; Spasoff, R.A.; Canadian Apheresis Study Group (August 8, 1991). "Comparison of Plasma Exchange with Plasma Infusion in the Treatment of Thrombotic Thrombocytopenic Purpura". New England Journal of Medicine. 325 (6): 393–397. doi:10.1056/nejm199108083250604. PMID 2062330.
- Knöbl, P. (2014). "Inherited and Acquired Thrombotic Thrombocytopenic Purpura (TTP) in Adults". Semin Thromb Hemost. 40 (4): 493–502. doi:10.1055/s-0034-1376883. PMID 24802084.
- Schiviz, A.; Wuersch, K.; Piskernik, C.; Dietrich, B.; Hoellriegl, W.; Rottensteiner, H.; Scheiflinger, F.; Schwarz, H.P.; Muchitsch, E. (2012). "A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13". Blood. 119 (25): 6128–6135. doi:10.1182/blood-2011-09-380535. PMID 22529289.
- Holz, J-B. (2012). "The TITAN trial – Assessing the efficacy and safety of an anti-von Willebrand factor nanobody in patients with acquired thrombotic thrombocytopenic purpura". Transfusion and Apheresis Science. 46 (3): 343–346. doi:10.1016/j.transci.2012.03.027. PMID 22475545.
- Mansouri Taleghani, M.; von Krogh, A-S.; Fujimura, Y.; George, J.N.; Hrachovinova, I.; Knöbl, P.N.; Quist-Paulsen, P.; Schneppenhiem, R.; Lämmle, B.; Kremer Hovinga, J.A. (May 29, 2013). "Hereditary thrombotic thrombocytopenic purpura and the hereditary TTP registry". Hamostaseologie. 33 (2).
- Terell, D.R.; Williams, L.A.; Vesely, S.K.; Lämmle, B.; Hovinga, J.A.K.; George, J.N. (July 2005). "The incidence of thrombotic thrombocytopenic purpura — hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency". Thrombosis and Haemostasis. 3 (7): 1432–1436. doi:10.1111/j.1538-7836.2005.01436.x. PMID 15978100.
- Reese, JA; Muthurajah, DS; Kremer Hovinga, JA; Vesely, SK; Terrell, DR; George, JN (October 2013). "Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired ADAMTS13 deficiency: comparison of incidence, demographic and clinical features". Pediatr Blood Cancer. 60 (10): 1676–1682. doi:10.1002/pbc.24612. PMID 23729372.
- Schneppheim, R.; Kremer Hovinga, J.A.; Becker, T.; Budde, U.; Karpman, D.; Brockhaus, W.; Harchovinová, I.; Korczowski, B.; Oyen, F.; Rittich, S.; von Rosen, J.; Tjonnfjord, G.E.; Pimanda, J.E.; Wienker, T.F.; Lämmle, B. (July 2006). "A common origin of the 4143insA ADAMtS13 mutation". Thrombosis and Haemostasis: 3–6.
- Upshaw, J.D. Jr. (June 15, 1978). "Congenital Deficiency of a Factor in Normal Plasma that Reverses Microangiopathic Heolysis and Thrombocytopenia". New England Journal of Medicine. 298 (24): 1350–1352. doi:10.1056/NEJM197806152982407. PMID 651994.
- Rennard, S.; Abe, S. (February 15, 1979). "Decreased cold-insoluble globulin in congenital thrombocytopenia (Upshaw-Schulman syndrome)". New England Journal of Medicine. 300 (7): 368. doi:10.1056/nejm197902153000717. PMID 759902.
- Tsai, H. (May 15, 1996). "Physiologic Cleavage of von Willebrand Factor by a Plasma Protease Is Dependent on Its Conformation and Requires Calcium Ion". Blood. 87 (10): 4235–4244. doi:10.1182/blood.V87.10.4235.bloodjournal87104235. PMID 8639782.
- Furlan, M.; Robles, R.; Solenthaler, M.; Wassmer, M.; Sandoz, P.; Lämmle, B. (May 1, 1997). "Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura". Blood. 89 (9): 3097–3103. doi:10.1182/blood.V89.9.3097. PMID 9129011.
External links
| Classification | |
|---|---|
| External resources |