Vicriviroc
Vicriviroc, previously named SCH 417690 and SCH-D, is a pyrimidine CCR5 entry inhibitor of HIV-1. It was developed by the pharmaceutical company Schering-Plough. Merck decided to not pursue regulatory approval for use in treatment-experienced patients because the drug did not meet primary efficacy endpoints in late stage trials. Clinical trials continue in patients previously untreated for HIV.
 | |
| Names | |
|---|---|
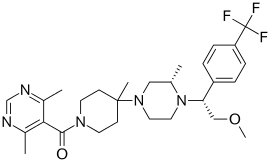
| IUPAC name
5-({4-[(3S)-4-{2-methoxy-1-[4-(trifluoromethyl)phenyl]ethyl}-3-methylpiperazin-1-yl]-4-methylpiperidin-1-yl}carbonyl)-4,6-dimethylpyrimidine | |
| Identifiers | |
| |
3D model (JSmol) |
|
| ChEMBL | |
| ChemSpider | |
| MeSH | Vicriviroc |
PubChem CID |
|
| UNII |
|
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C28H38F3N5O2 | |
| Molar mass | 533.629 g/mol |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
HIV-1 background
The mechanisms of a number of available anti-HIV drugs prevent either viral reverse transcriptase enzyme or protease enzyme, allowing the virus to enter the cell before these drugs take effect. However, CCR5 inhibitors such as vicriviroc, as well as other entry inhibitors of HIV-1, inhibit the initial stages of the virus life cycle.[1]
HIV-1 entry
HIV binds to and fuses with the target T-cells or macrophages with the help of gp120 and gp41, the only two proteins that are currently known to be exhibited on the surface of the viral envelope.[2] One molecule of each protein associates noncovalently with the other on the viral membrane, and three of these units aggregate to form the gp120/gp41 heterotrimer, which traps the gp41 in a conformationally metastable state.[2]
Membrane fusion begins with the binding of gp120 to CD4, a glycoprotein which is expressed on the surface of the target cell.[1] Upon binding, gp120 undergoes a conformational change, which causes the formation of the coreceptor binding site on gp120.[2] All strains of HIV-1 use one of two coreceptors: CCR5 or CXCR4; coreceptor specificity will be described below. Once gp120 binds to the coreceptor, gp41 undergoes a conformational change that releases it from its once-metastable position.[2] This change causes the hydrophobic N-terminus of the gp41 protein, also known as the fusion domain, to insert into the host cell membrane and anchor the virus into place.[1][2] The insertion of gp41 into the target cell causes a subtle rearrangement in the gp41 protein that brings together two trimeric coiled coils, HR1 and HR2, to form a six-helix bundle.[2] The bundle allows the viral and cellular membranes to approximate and eventually fuse together, leading to the release of the viral genome into the cytoplasm of the target cell.[2]
Coreceptors and tropism
The two coreceptors involved in the entry of HIV-1, CCR5 and CXCR4, belong to the larger family of 7-transmembrane segment (7TM) G-protein coupled receptors.[2] HIV-1 can thus be classified according to specificity for one coreceptor or the other. R5 virus, also known as M-tropic HIV-1, targets macrophages and uses CCR5 as the coreceptor. X4 virus, or T-tropic HIV-1, targets T-cells and uses CXCR4 as the coreceptor. Dual-tropic strains of HIV-1, which utilize both receptors, also exist.[1] Selectivity for one coreceptor or the other is especially dependent upon the V3 loop, a highly variable and structurally flexible region of gp120 that is composed of approximately 35 amino acids. Tropism can be predicted through the 11/25 method, which looks for basic amino acids at positions 11 and 25 in the V3 loop and suggests the presence of an X4 virus.[2]
Coreceptor usage, however, can change throughout the course of infection. 90% of patients in early phases of HIV-1 infection have R5 virus. However, after five years of infection, about 50% of all patients have detectable amounts of X4 virus.[2] Causes for this switch are currently unclear. However, viral changes from CCR5 to CXCR4 coreceptor usage have been associated with a faster rate of CD4+ T-cell loss, rapid viral progression, and an increased rate of development of AIDS and death.[1][2]
CCR5-Δ32
A focus on the CCR5 co-receptor as a potential target for anti-HIV medications began in 1996 with the discovery of CCR5-Δ32, or CCR5 delta-32, a mutational variant of the CCR5 coding gene.[1] The deletion of 32 base pairs in this gene results in nonfunctional CCR5 receptors.[1] While the frequency of this mutation within Caucasian populations is 0.0808, people of African or Asian descent do not appear to possess this allele.[1] Δ32 homozygotes, or individuals who possess two copies of the Δ32 variant, have no functional CCR5 receptors and are consequently highly resistant to HIV infection.[1] Individuals who inherit one copy of Δ32 variant and one copy of the normal CCR5 gene are CCR5 heterozygotes.[1] Δ32 heterozygotes are still susceptible to HIV-1 infection, but the progression of the disease is significantly delayed compared to those with two normal copies of the CCR5 gene. CCR5 antagonists have been developed which cause deformation in the CCR5 co-receptor, leading to the cell's failure to bind with the HIV gp120 protein.[1]
SCH-C and vicriviroc
In 2001, Schering-Plough developed one of the first small molecule CCR5 antagonists, SCH-C or SCH 351125, which inhibited replication of a number of HIV-1 isolates that used CCR5 as a coreceptor for binding.[3] However, SCH-C caused a modest but dose-dependent prolongation of the corrected cardiac QT interval (QTc),[4] leading to examination of alternative compounds whose antiviral and pharmacokinetic properties exceeded those of first-generation compounds like SCH-C. Vicriviroc was discovered in high-throughput screening and structure-activity relationships (SAR) analysis.[5] When compared with SCH-C, vicriviroc consistently and more actively inhibits viral replication, binds with higher affinity to CCR5 than SCH-C, and possesses a lower affinity for the human ether-a-go-go-related gene transcript ion channel, which may suggest a lower risk of cardiac effects.[4]
Method of action
Vicriviroc is a noncompetitive allosteric antagonist of CCR5.[6] It is orally administered and, because it is effective at nanomolar concentrations, it can be administered once daily.[1][6] Vicriviroc binds to a small hydrophobic pocket between the transmembrane helices near the extracellular surface of the CCR5 receptor.[1] Binding to this pocket induces a conformational change of the extracellular segment of CCR5 and prevents binding of gp120 to the target cell, consequently preventing the virus from entering the target cell at all.
Specific binding interactions between CCR5 and vicriviroc were first described in 2008.[7] Specifically, the trifluoromethyl phenyl group of vicriviroc may interact strongly with I198 residue on the fifth transmembrane helix (TM5) of CCR5 through hydrophobic interactions. Additionally, electrostatic interactions may form between the positively charged tertiary nitrogen group of vicriviroc and the hydrophilic region provided by E238 residue on TM7 of CCR5. Other strong interactions predicted by the group include the Y108 residue on TM3 and Y251 on TM6.
Clinical trials
Currently, vicriviroc is undergoing critical trials. Vicriviroc demonstrated a significant decrease of HIV RNA in R5-infected subjects. The mean decline from baseline of HIV RNA achieved 1.5 log10 or greater in all treatment groups (10, 25, 50 mg, b.i.d.) in a 14-day monotherapy trial in HIV-infected adults.[8]
A phase II trial in treatment-naïve HIV-1 infected subjects was discontinued after the rate of virologic relapse in those subjects who were administered vicriviroc increased compared to control subjects; however, further investigations suggest that the administered dosage of vicriviroc may have been too low.[2] A new Phase II trial of treatment-naive HIV-1 patients is currently underway.
A 48-week phase II trial (ACTG5211) examining the safety and efficacy of 5, 10, and 15 mg doses of vicriviroc found that patients in the 10 mg and 15 mg vicriviroc treatment groups achieved a median decrease in viral load of 1.92 and 1.44 (log10 copies/mL) and a median increase in CD4 cell count of 130 and 96 (cell/uL) from baseline, respectively. More patients in the vicriviroc groups had undetectable virus at 48 weeks (HIV-1 RNA <400/<50 copies/ml) compared to those in the placebo group (57/37 percent and 43/27 percent vs. 14/11 percent, respectively).[9]
The results from a 48-week phase II trial (VICTOR-E1) examining administration of 20 or 30 mg dosages of vicriviroc in addition to > 3-drug optimized background therapy (OBT) regimen that included a ritonavir-boosted protease inhibitor were reported in February 2008. Investigators concluded that, “‘Vicriviroc 30 or 20 mg once daily plus ritonavir-containing OBT provided sustained viral suppression in treatment-experienced subjects and increased CD4 cell counts regardless of the number of active drugs in OBT.’"[10]
As of May 2008, two phase III trials (VICTOR-E3 and VICTOR E4) in treatment-experienced patients were initiated.[11] The late stage clinical trials by did not meet their primary efficacy endpoints and Merck has decided as of January 2010 to not pursue regulatory approval for the drug.[12][13]
Concerns surrounding CCR5 antagonists
Available clinical trial data suggest that a new method of combating HIV-1 may be found in CCR5 antagonists. Studies into vicriviroc are ongoing, and another CCR5 antagonist, maraviroc, is currently on the market. However, concerns have arisen regarding the use of CCR5 antagonists as anti-HIV treatments because such drugs may facilitate the emergence of resistant strains of HIV-1. Two possibilities for such resistance have been hypothesized: HIV-1 may evolve to use only CXCR4 as the coreceptor; or HIV-1 may mutate in such a way that it is still able to interact with CCR5, despite the presence of a receptor antagonist.[1] In fact, maraviroc-resistant variants of HIV-1 have already been generated in vitro by mutating residues in the V3 loop of gp120.[2]
References
- Idemyor V (2005). "Human immunodeficiency virus (HIV) entry inhibitors (CCR5 specific blockers) in development: are they the next novel therapies?". HIV Clin Trials. 6 (5): 272–7. doi:10.1310/979L-39QP-NC9G-WFTF. PMID 16306033. S2CID 31384269.
- Tsibris A (2007). "Update On CCR5 Inhibitors: Scientific Rationale, Clinical Evidence, and Anticipated Uses". Physicians’ Research Network. Retrieved 2008-05-11.
- Palani A, Shapiro S, Clader JW, Greenlee WJ, Cox K, Strizki J, Endres M, Baroudy BM (2001). "Discovery of 4-[(Z)-(4-bromophenyl)- (ethoxyimino)methyl]-1'-[(2,4-dimethyl-3- pyridinyl)carbonyl]-4'-methyl-1,4'- bipiperidine N-oxide (SCH 351125): an orally bioavailable human CCR5 antagonist for the treatment of HIV infection". J Med Chem. 44 (21): 3339–42. doi:10.1021/jm015526o. PMID 11585437.
- Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, Hipkin RW, Chou CC, Pugliese-Sivo C, Xiao Y, Tagat JR, Cox K, Priestley T, Sorota S, Huang W, Hirsch M, Reyes GR, Baroudy BM (2005). "Discovery and Characterization of Vicriviroc (SCH 417690), a CCR5 Antagonist with Potent Activity against Human Immunodeficiency Virus Type 1". Antimicrobial Agents and Chemotherapy. 49 (12): 4911–4919. doi:10.1128/AAC.49.12.4911-4919.2005. PMC 1315929. PMID 16304152.
- Tagat JR, McCombie SW, Nazareno D, Labroli MA, Xiao Y, Steensma RW, Strizki JM, Baroudy BM, Cox K, et al. (2004). "Piperazine-based CCR5 antagonists as HIV-1 inhibitors. IV. Discovery of 1-[(4,6-dimethyl-5-pyrimidinyl)carbonyl]- 4-[4-[2-methoxy-1(R)-4-(trifluoromethyl)phenyl]ethyl-3(S)-methyl-1-piperazinyl]- 4-methylpiperidine (Sch-417690/Sch-D), a potent, highly selective, and orally bioavailable CCR5 antagonist". J Med Chem. 47 (10): 2405–8. doi:10.1021/jm0304515. PMID 15115380.
- AIDSinfo (2007). "Vicriviroc maleate". NIH. Retrieved 2008-05-11.
- Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, Dioszegi M (2008). "Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists". Mol. Pharmacol. 73 (3): 789–800. doi:10.1124/mol.107.042101. PMID 18096812. S2CID 16267853.
- Schürmann D, et al. (2007). "Antiviral activity, pharmacokinetics and safety of vicriviroc, an oral CCR5 antagonist, during 14-day monotherapy in HIV-infected adults". AIDS. 21 (10): 1293–9. doi:10.1097/QAD.0b013e3280f00f9f. PMID 17545705. S2CID 6752651.
- Baker R (2007). "Safety and Efficacy of Experimental CCR5 Antagonist Vicriviroc in Treatment-experienced HIV Patients: 48 week Results of ACTG 5211". HIVandHepatitis.com. Archived from the original on 23 August 2007. Retrieved 2008-05-11.
- Highleyman L (2008). "CCR5 Antagonist Vicriviroc Shows Continued Benefits and Good Tolerability at 48 Weeks: VICTOR-E1 Trial". HIVandHepatitis.com. Retrieved 2008-05-11.
- Baker R (2008). "Schering-Plough Opens Enrollment to 2 Phase III Trials of the Experimental CCR5 Antagonist Vicriviroc". HIVandHepatitis.com. Archived from the original on 2007-11-12. Retrieved 2008-05-11.
- Loftus P (2010). "Merck Won't Seek FDA Approval For HIV Drug". Wall Street Journal. wsj.com. Retrieved 2010-01-21.
- Pierson R (2010-01-20). "Merck HIV drug from Schering merger fails trials". reuters.com. Retrieved 2010-01-21.