Aceruloplasminemia
Aceruloplasminemia is a rare autosomal recessive disorder[2] in which the liver can not synthesis the protein ceruloplasmin properly, which is needed to transport copper around the blood. Copper deficiency in the brain results in neurological problems that generally appear in adulthood and worsen over time. .[3]
| Aceruloplasminemia | |
|---|---|
| Other names | Ceruloplasmin deficiency[1] |
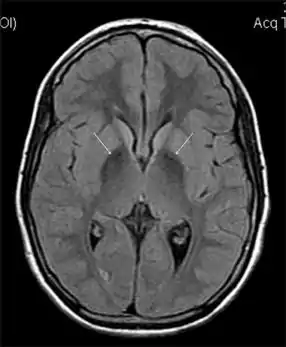 | |
| MRI hypointensity (white arrows) in the globus pallidi that indicates the presence of iron deficiencies which would be seen in a diagnosis of Aceruloplasminemia | |
| Specialty | Medical genetics |
Aceruloplasminemia has been seen worldwide, but its overall prevalence is unknown. Studies in Japan have estimated that approximately 1 in 2 million adults in this population are affected.[4]
Aceruloplasminemia belongs to the group of genetic disorders called neurodegeneration with brain iron accumulation (NBIA).
Signs and symptoms
Patients with aceruloplasminemia develop a variety of movement problems. They may experience dystonia of the head and neck, resulting in repetitive movements and contortions. Other involuntary movements may also occur, such as tremors, chorea, blepharospasms, and grimacing. Affected individuals may also experience ataxia, the lack of coordination of muscle movements. Some develop psychiatric problems and midlife dementia.[4] The type of neurological disruption corresponds to associated regions of iron deposition in the brain and liver.[5]
In addition to neurological problems, affected individuals may have diabetes mellitus caused by iron damage to cells in the pancreas that make insulin. This impairs blood sugar regulation and leads to the signs and symptoms of diabetes.[4]
Iron accumulation in the tissues and organs results in a corresponding iron deficiency in the blood, leading to anemia. Anemia and diabetes usually occur by the time an affected person is in his or her twenties.[4]
Affected individuals also experience retinal degeneration caused by excess iron. The changes result in small opaque spots and areas of atrophy around the edges of the retina. These abnormalities usually do not affect vision but can be observed during an eye examination.[4]
Cause
Aceruloplasminemia is caused by a mutation (a five-base pair insertion in exon 7[3]) in the CP gene, which provides instructions for making a protein called ceruloplasmin, a protein involved in iron transport and processing. Ceruloplasmin helps move iron from the organs and tissues of the body and prepares it for incorporation into a molecule called transferrin, which transports it to red blood cells to help carry oxygen. The CP gene mutation results in the production of ceruloplasmin protein that is unstable or nonfunctional by altering the open reading frame such that the amino acid ligands in the essential carboxyl terminal region are eliminated.[3] When ceruloplasmin is unavailable, transport of iron out of the body's tissues is impaired. The resulting iron accumulation damages cells in those tissues, leading to neurological dysfunction and other health problems.[4][6]
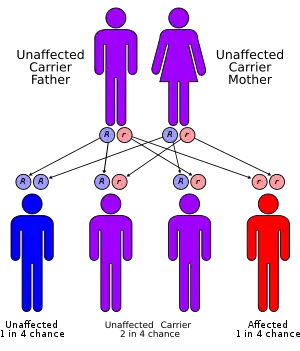
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene have the mutation. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.[4]
Diagnosis
Diagnosis of this disorder depends on blood tests demonstrating the absence of serum ceruloplasmin, combined with low serum copper concentration, low serum iron concentration, high serum ferritin concentration, or increased hepatic iron concentration. MRI scans can also confirm a diagnosis; abnormal low intensities can indicate iron accumulation in the brain.[5]
Prevention
Children of affected individuals are obligate carriers for aceruloplasminemia. If the CP mutations has been identified in a related individual, prenatal testing is recommended. Siblings of those affected by the disease are at a 25% of aceruloplasminemia. In asymptomatic siblings, serum concentrations of hemoglobin and hemoglobin A1c should be monitored.[5]
To prevent the progression of symptoms of the disease, annual glucose tolerance tests beginning in early teen years to evaluate the onset of diabetes mellitus. Those at risk should avoid taking iron supplements.[5]
Treatment
Treatment includes the use of iron chelating agents (such as desferrioxamine) to lower brain and liver iron stores, and to prevent progression of neurologic symptoms. This, combined with fresh-frozen human plasma (FFP) works effectively in decreasing liver iron content. Repetitive use of FFP can even improve neurologic symptoms. Antioxidants such as vitamin E can be used simultaneously to prevent tissue damage to the liver and pancreas.[5]
References
- "Aceruloplasminemia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 16 April 2019.
- Harris, Zl; Takahashi, Y; Miyajima, H; Serizawa, M; Macgillivray, Rt; Gitlin, Jd (March 1995). "Aceruloplasminemia: molecular characterization of this disorder of iron metabolism". Proceedings of the National Academy of Sciences of the United States of America. 92 (7): 2539–43. Bibcode:1995PNAS...92.2539H. doi:10.1073/pnas.92.7.2539. ISSN 0027-8424. PMC 42253. PMID 7708681.
- Harris, ZL; Klomp, LW; Gitlin, JD (May 1998). "Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis". The American Journal of Clinical Nutrition. 67 (5 Suppl): 972S–977S. doi:10.1093/ajcn/67.5.972S. PMID 9587138.
- "Aceruloplasminemia". Genetics Home Reference. U.S. National Library of Medicine. 10 February 2014. Retrieved 11 February 2014.
- Miyajima H (1993). Pagon RA; Adam MP; Ardinger HH; Bird TD; Dolan CR; Fong CT; Smith RJH; Stephens K (eds.). "Aceruloplasminemia". GeneReviews. PMID 20301666. Retrieved 2014-02-12.
- Yoshida, K; Furihata, K; Takeda, S; Nakamura, A; Yamamoto, K; Morita, H; Hiyamuta, S; Ikeda, S; Shimizu, N; Yanagisawa, N (March 1995). "A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans". Nature Genetics. 9 (3): 267–72. doi:10.1038/ng0395-267. PMID 7539672.
External links
| Classification |
|---|