Hemosiderosis
Hemosiderosis is a form of iron overload disorder resulting in the accumulation of hemosiderin.
| Hemosiderosis | |
|---|---|
| Other names | Haemosiderosis |
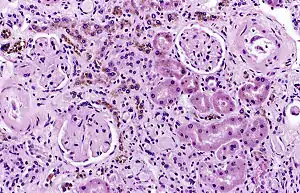 | |
| Image of a kidney viewed under a microscope. The brown areas contain hemosiderin | |
| Specialty | Hematology |
Types include:
Hemosiderin deposition in the lungs is often seen after diffuse alveolar hemorrhage, which occurs in diseases such as Goodpasture's syndrome, granulomatosis with polyangiitis, and idiopathic pulmonary hemosiderosis. Mitral stenosis can also lead to pulmonary hemosiderosis. Hemosiderin collects throughout the body in hemochromatosis. Hemosiderin deposition in the liver is a common feature of hemochromatosis and is the cause of liver failure in the disease. Selective iron deposition in the beta cells of pancreatic islets leads to diabetes[4][2] due to distribution of transferrin receptor on the beta cells of islets[3] and in the skin leads to hyperpigmentation. Hemosiderin deposition in the brain is seen after bleeds from any source, including chronic subdural hemorrhage, cerebral arteriovenous malformations, cavernous hemangiomata. Hemosiderin collects in the skin and is slowly removed after bruising; hemosiderin may remain in some conditions such as stasis dermatitis. Hemosiderin in the kidneys has been associated with marked hemolysis and a rare blood disorder called paroxysmal nocturnal hemoglobinuria.
Hemosiderin may deposit in diseases associated with iron overload. These diseases are typically diseases in which chronic blood loss requires frequent blood transfusions, such as sickle cell anemia and thalassemia, though beta thalassemia minor has been associated with hemosiderin deposits in the liver in those with non-alcoholic fatty liver disease independent of any transfusions.[5][6]
Iron overload occurs when iron intake is increased over a sustained period of time due to regular transfusion of whole blood and red cells or because of increased absorption of iron through the gastrointestinal tract (GI).
Both these phenomena occur in thalassaemias, with blood transfusion therapy being the major cause of iron overload in thalassaemia major and increased GI absorption being more important in patients with intermedia thalassaemia who are not frequently transfused.
Each unit of blood contains about 200mg iron. After 50 units have been transfused, or earlier in children, siderosis develops, with increased pigmentation of skin exposed to light and susceptibility to infection, reduced growth and delayed sexual development and puberty(24). The recommended red cell transfusion scheme for patients with β-thalassaemia amounts to 116-232 mg iron per Kg weight on an annual basis (0.32-0.64 mg/Kg/day).
The human body lacks a mechanism to excrete excess iron. Iron accumulation is toxic to many tissues, causing heart failure, cirrhosis, liver cancer, growth retardation and endocrine abnormalities. In the absence of regular iron chelation therapy, the iron loading rates vary. Monitoring of transfusion iron overload is essential for effective and safe iron chelation tailored to the individual’s specific needs.
Serum ferritin (SF) measured at least every 3 months (the currently accepted target value is between 500-1000 mg/L) should also be evaluated along with the liver iron concentration (LIC) assessed using a validated and standardized MRI technique and myocardial iron as measured by MRI-based methods with specific software T2*.
For monitoring of transfusion iron overload, other organ function and iron-mediated damage, surveillance of the patient for diabetes, hypothyroidism, hypoparathyroidism and hypoponadotropic hypogonadism is recommended.
Diagnosis
There are several methods available for diagnosing and monitoring hemosiderosis including:
Serum ferritin is a low cost, readily available, and minimally invasive method for assessing body iron stores. However, the major problem with using it as an indicator of hemosiderosis is that it can be elevated in a range of other medical conditions unrelated to iron levels including infection, inflammation, fever, liver disease, renal disease and cancer.
While liver biopsies provide a direct measure of liver iron concentration, the small sample size relative to the size of the liver can lead to sampling errors given the heterogeneity of iron concentration within the liver. Furthermore, the invasive nature of liver biopsy and the associated risks of complications (which can range from pain, haemorrhage, gallbladder perforation and other morbidities through to death in approx 1 in 10,000 cases) prevent it being used as a regular monitoring tool.
MRI is emerging as an alternative method for measuring liver iron loading because it is non-invasive, safer and generally cheaper to perform than liver biopsy; does not suffer from problems with sampling variability; and can be used more frequently than performing liver biopsies.[7]
Treatment
Treatment for hemosiderin focuses on limiting the effects of the underlying disease leading to continued deposition. In hemochromatosis, this entails frequent phlebotomy granulomatosis, immune suppression is required. Limiting blood transfusions and institution of iron chelation therapy when iron overload is detected are important when managing sickle-cell anemia and other chronic hemolytic anemias.
The aims of iron chelation therapy include (a) prevention therapy in order to minimize the risk of onset of iron-mediated complications, (b) rescue therapy for the removal of storage iron and (c) emergency therapy if heart failure develops or if there is a downward trend of left ventricular (LV) function that requires hospitalisation using continuous intravenous desferrioxamine (DFO), possibly combined with deferiprone (DFP). It aims to balance the rate of iron accumulation from blood transfusion by increasing iron excretion in urine and in faeces with chelators.
There are currently three licensed iron chelators, DFO, DFP and Deferasirox (DFX). The Guide for the Management of Transfusion Dependent Thalassaemia (TDT) issued by the Thalassaemia International Federation (TIF Publication No23, 2017) contains details of dose and regimen adjustment of iron chelation therapy, adherence to therapy and use of combination therapies as well as monitoring of chelation therapy in special circumstances such as pregnancy, renal impairment and summary recommendations.
See also
References
- Lu JP, Hayashi K (1995). "Transferrin receptor distribution and iron deposition in the hepatic lobule of iron-overloaded rats". Pathol. Int. 45 (3): 202–6. doi:10.1111/j.1440-1827.1995.tb03443.x. PMID 7787990. S2CID 22932153.
- Lu JP, Hayashi K (1994). "Selective iron deposition in pancreatic islet B cells of transfusional iron-overloaded autopsy cases". Pathol. Int. 44 (3): 194–9. doi:10.1111/j.1440-1827.1994.tb02592.x. PMID 8025661. S2CID 25357672.
- Lu JP, Hayashi K, Okada S, Awai M (1991). "Transferrin receptors and selective iron deposition in pancreatic B cells of iron-overloaded rats". Acta Pathol Jpn. 41 (9): 647–52. doi:10.1111/j.1440-1827.1991.tb02787.x. PMID 1776464. S2CID 42444122.
- Valenti, Luca; Canavesi, Elena; Galmozzi, Enrico; Dongiovanni, Paola; Rametta, Raffaela; Maggioni, Paolo; Maggioni, Marco; Fracanzani, Anna Ludovica; Fargion, Silvia (2010). "Beta-globin mutations are associated with parenchymal siderosis and fibrosis in patients with non-alcoholic fatty liver disease". Journal of Hepatology. 53 (5): 927–33. doi:10.1016/j.jhep.2010.05.023. PMID 20739079.
- Stickel, Felix; Hampe, Jochen (2010). "Dissecting the evolutionary genetics of iron overload in non-alcoholic fatty liver disease". Journal of Hepatology. 53 (5): 793–4. doi:10.1016/j.jhep.2010.06.010. PMID 20739088.
- St. Pierre, T. G.; Clark, PR; Chua-Anusorn, W; Fleming, AJ; Jeffrey, GP; Olynyk, JK; Pootrakul, P; Robins, E; Lindeman, R (2005). "Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance". Blood. 105 (2): 855–61. doi:10.1182/blood-2004-01-0177. PMID 15256427.
8. The Guide for the Management of Transfusion Dependent Thalassaemia (TDT) 3rd edition, editors Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V, published and issued by the Thalassaemia International Federation (TIF Publication No23, 2017)