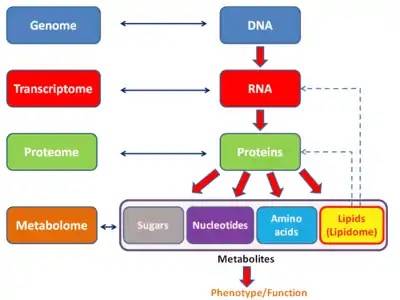
Proteome
The proteome is the entire set of proteins that is, or can be, expressed by a genome, cell, tissue, or organism at a certain time. It is the set of expressed proteins in a given type of cell or organism, at a given time, under defined conditions. Proteomics is the study of the proteome.
Systems
The term has been applied to several different types of biological systems.
A cellular proteome is the collection of proteins found in a particular cell type under a particular set of environmental conditions such as exposure to hormone stimulation.
It can also be useful to consider an organism's complete proteome, which can be conceptualized as the complete set of proteins from all of the various cellular proteomes. This is very roughly the protein equivalent of the genome.
The term "proteome" has also been used to refer to the collection of proteins in certain sub-cellular biological systems. For example, all of the proteins in a virus can be called a viral proteome. All of the proteins in a mitochondrion make up the mitochondrial proteome which has generated its own field of study mitoproteomics.[1]
Importance in cancer
The proteome can be used in order to comparatively analyze different cancer cell lines. Proteomic studies have been used in order to identify the likelihood of metastasis in bladder cancer cell lines KK47 and YTS1 and were found to have 36 unregulated and 74 down regulated proteins.[2] The differences in protein expression can help identify novel cancer signaling mechanisms.
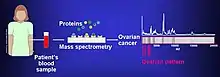
Biomarkers of cancer have been found by mass spectrometry based proteomic analyses. The use of proteomics or the study of the proteome is a step forward in personalized medicine to tailor drug cocktails to the patient's specific proteomic and genomic profile.[3] The analysis of ovarian cancer cell lines showed that putative biomarkers for ovarian cancer include "α-enolase (ENOA), elongation factor Tu, mitochondrial (EFTU), glyceraldehyde-3-phosphate dehydrogenase (G3P), stress-70 protein, mitochondrial (GRP75), apolipoprotein A-1 (APOA1), peroxiredoxin (PRDX2) and annexin A (ANXA)".[4]
Comparative proteomic analyses of 11 cell lines demonstrated the similarity between the metabolic processes of each cell line; 11,731 proteins were completely identified from this study. Housekeeping proteins tend to show greater variability between cell lines.[5]
Resistance to certain cancer drugs is still not well understood. Proteomic analysis has been used in order to identify proteins that may have anti-cancer drug properties, specifically for the colon cancer drug irinotecan.[6] Studies of adenocarcinoma cell line LoVo demonstrated that 8 proteins were unregulated and 7 proteins were down-regulated. Proteins that showed a differential expression were involved in processes such as transcription, apoptosis and cell proliferation/differentiation among others.
The proteome in bacterial systems
Proteomic analyses have been performed in different kinds of bacteria to assess their metabolic reactions to different conditions. For example, in bacteria such as Clostridium and Bacillus, proteomic analyses were used in order to investigate how different proteins help each of these bacteria spores germinate after a prolonged period of dormancy.[7] In order to better understand how to properly eliminate spores, proteomic analysis must be performed.
History
Marc Wilkins coined the term proteome [8] in 1994 in a symposium on "2D Electrophoresis: from protein maps to genomes" held in Siena in Italy. It appeared in print in 1995,[9] with the publication of part of his PhD thesis. Wilkins used the term to describe the entire complement of proteins expressed by a genome, cell, tissue or organism.
Size and contents
Assuming a one-to-one correspondence between genes and proteins would mean that there are at least 20,000 proteins corresponding to roughly 20,000 genes for humans. The proteome can be larger than the genome, especially in eukaryotes, as more than one protein can be produced from one gene due to alternative splicing (e.g. human proteome consists 92,179 proteins out of which 71,173 are splicing variants).[10] On the other hand, not all genes are translated to proteins, and many known genes encode only RNA which is the final functional product. Moreover, complete proteome size varies depending on the kingdom of life. For instance, eukaryotes, bacteria, archaea and viruses have on average 15,145, 3,200, 2,358 and 42 proteins respectively encoded in their genomes.[11]
The Plasma Proteome database contains information on 10,500 blood plasma proteins. Because the range in protein contents in plasma is very large, it is difficult to detect proteins that tend to be scarce when compared to abundant proteins. There is an analytical limit that may possibly be a barrier for the detections of proteins with ultra low concentrations.[12]
There are different factors that are used to analyze the proteome. First, the protein width is determined by the different protein types and the protein depth is determined by the number of protein copies in particular tissues.[12] There are different factors that can add variability to proteins. SAPs (single amino acid polymorphisms) and nsSNPs non-synonymous single nucleotide polymorphisms are key elements that can lead to different "protein species" or "proteomorphs".
The term dark proteome coined by Perdigão and colleagues, defines regions of proteins that have no detectable sequence homology to other proteins of known three-dimensional structure and therefore cannot be modeled by homology. For 546,000 Swiss-Prot proteins, 44–54% of the proteome in eukaryotes and viruses was found to be "dark", compared with only ∼14% in archaea and bacteria.[13]
Currently, several projects aim to map the human proteome, including the Human Proteome Map, ProteomicsDB and The Human Proteome Project (HPP). Much like the human genome project, these projects seek to find and collect evidence for all predicted protein coding genes in the human genome. The Human Proteome Map currently (October 2020) claims 17,294 proteins and ProteomicsDB 15,479, using different criteria. On October 16, 2020, the HPP published a high-stringency blueprint [14] covering more than 90% of the predicted protein coding genes. Proteins are identified from a wide range of fetal and adult tissues and cell types, including hematopoietic cells. Databases such as neXtprot and UniProt are central resources for human proteomic data.
Methods to study the proteome

Analyzing proteins proves to be more difficult than analyzing nucleic acid sequences. While there are only 4 nucleotides that make up DNA, there are at least 20 different amino acids that can make up a protein. Additionally, there is currently no known high throughput technology to make copies of a single protein. Numerous methods are available to study proteins, sets of proteins, or the whole proteome. In fact, proteins are often studied indirectly, e.g. using computational methods and analyses of genomes. Only a few examples are given below.
Separation techniques and electrophoresis
Proteomics, the study of the proteome, has largely been practiced through the separation of proteins by two dimensional gel electrophoresis. In the first dimension, the proteins are separated by isoelectric focusing, which resolves proteins on the basis of charge. In the second dimension, proteins are separated by molecular weight using SDS-PAGE. The gel is stained with Coomassie Brilliant Blue or silver to visualize the proteins. Spots on the gel are proteins that have migrated to specific locations.
Mass spectrometry

Mass spectrometry is one of the key methods to study the proteome.[15] Some important mass spectrometry methods include Orbitrap Mass Spectrometry, MALDI (Matrix Assisted Laser Desorption/Ionization), and ESI (Electrospray Ionization). Peptide mass fingerprinting identifies a protein by cleaving it into short peptides and then deduces the protein's identity by matching the observed peptide masses against a sequence database. Tandem mass spectrometry, on the other hand, can get sequence information from individual peptides by isolating them, colliding them with a non-reactive gas, and then cataloguing the fragment ions produced.[16]
In May 2014, a draft map of the human proteome was published in Nature.[17] This map was generated using high-resolution Fourier-transform mass spectrometry. This study profiled 30 histologically normal human samples resulting in the identification of proteins coded by 17,294 genes. This accounts for around 84% of the total annotated protein-coding genes.
Chromatography
Liquid chromatography is an important tool in the study of the proteome. It allows for very sensitive separation of different kinds of proteins based on their affinity for a matrix. Some newer methods for the separation and identification of proteins include the use of monolithic capillary columns, high temperature chromatography and capillary electrochromatography.[18]
Blotting
Western blotting can be used in order to quantify the abundance of certain proteins. By using antibodies specific to the protein of interest, it is possible to probe for the presence of specific proteins from a mixture of proteins.
Protein complementation assays and interaction screens
Protein-fragment complementation assays are often used to detect protein–protein interactions. The yeast two-hybrid assay is the most popular of them but there are numerous variations, both used in vitro and in vivo. Pull-down assays are a method to determine what kinds of proteins a protein interacts with.[19]
See also
- Metabolome
- Cytome
- Bioinformatics
- List of omics topics in biology
- Plant Proteome Database
- Transcriptome
- Interactome
- Human Proteome Project
- BioPlex
References
- Gómez-Serrano, M (November 2018). "Mitoproteomics: Tackling Mitochondrial Dysfunction in Human Disease". Oxid Med Cell Longev. 2018: 1435934. doi:10.1155/2018/1435934. PMC 6250043. PMID 30533169.
- Yang, Ganglong; Xu, Zhipeng; Lu, Wei; Li, Xiang; Sun, Chengwen; Guo, Jia; Xue, Peng; Guan, Feng (2015-07-31). "Quantitative Analysis of Differential Proteome Expression in Bladder Cancer vs. Normal Bladder Cells Using SILAC Method". PLOS ONE. 10 (7): e0134727. Bibcode:2015PLoSO..1034727Y. doi:10.1371/journal.pone.0134727. ISSN 1932-6203. PMC 4521931. PMID 26230496.
- An, Yao; Zhou, Li; Huang, Zhao; Nice, Edouard C.; Zhang, Haiyuan; Huang, Canhua (2019-05-04). "Molecular insights into cancer drug resistance from a proteomics perspective". Expert Review of Proteomics. 16 (5): 413–429. doi:10.1080/14789450.2019.1601561. ISSN 1478-9450. PMID 30925852. S2CID 88474614.
- Cruz, Isa N.; Coley, Helen M.; Kramer, Holger B.; Madhuri, Thumuluru Kavitah; Safuwan, Nur a. M.; Angelino, Ana Rita; Yang, Min (2017-01-01). "Proteomics Analysis of Ovarian Cancer Cell Lines and Tissues Reveals Drug Resistance-associated Proteins". Cancer Genomics - Proteomics. 14 (1): 35–51. doi:10.21873/cgp.20017. ISSN 1109-6535. PMC 5267499. PMID 28031236.
- Geiger, Tamar; Wehner, Anja; Schaab, Christoph; Cox, Juergen; Mann, Matthias (March 2012). "Comparative Proteomic Analysis of Eleven Common Cell Lines Reveals Ubiquitous but Varying Expression of Most Proteins". Molecular & Cellular Proteomics. 11 (3): M111.014050. doi:10.1074/mcp.M111.014050. ISSN 1535-9476. PMC 3316730. PMID 22278370.
- Peng, Xing-Chen; Gong, Feng-Ming; Wei, Meng; Chen, Xi; Chen, Ye; Cheng, Ke; Gao, Feng; Xu, Feng; Bi, Feng; Liu, Ji-Yan (December 2010). "Proteomic analysis of cell lines to identify the irinotecan resistance proteins". Journal of Biosciences. 35 (4): 557–564. doi:10.1007/s12038-010-0064-9. ISSN 0250-5991. PMID 21289438. S2CID 6082637.
- Chen, Yan; Barat, Bidisha; Ray, W. Keith; Helm, Richard F.; Melville, Stephen B.; Popham, David L. (2019-03-15). "Membrane Proteomes and Ion Transporters in Bacillus anthracis and Bacillus subtilis Dormant and Germinating Spores". Journal of Bacteriology. 201 (6). doi:10.1128/JB.00662-18. ISSN 0021-9193. PMC 6398275. PMID 30602489.
- Wilkins, Marc (Dec 2009). "Proteomics data mining". Expert Review of Proteomics. England. 6 (6): 599–603. doi:10.1586/epr.09.81. PMID 19929606. S2CID 207211912.
- Wasinger VC, Cordwell SJ, Cerpa-Poljak A, Yan JX, Gooley AA, Wilkins MR, Duncan MW, Harris R, Williams KL, Humphery-Smith I (1995). "Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium". Electrophoresis. 16 (1): 1090–94. doi:10.1002/elps.11501601185. PMID 7498152. S2CID 9269742.
- "UniProt: a hub for protein information". Nucleic Acids Research. 43 (D1): D204–D212. 2014. doi:10.1093/nar/gku989. ISSN 0305-1048. PMC 4384041. PMID 25348405.
- Kozlowski, LP (26 October 2016). "Proteome-pI: proteome isoelectric point database". Nucleic Acids Research. 45 (D1): D1112–D1116. doi:10.1093/nar/gkw978. PMC 5210655. PMID 27789699.
- Ponomarenko, Elena A.; Poverennaya, Ekaterina V.; Ilgisonis, Ekaterina V.; Pyatnitskiy, Mikhail A.; Kopylov, Arthur T.; Zgoda, Victor G.; Lisitsa, Andrey V.; Archakov, Alexander I. (2016). "The Size of the Human Proteome: The Width and Depth". International Journal of Analytical Chemistry. 2016: 7436849. doi:10.1155/2016/7436849. ISSN 1687-8760. PMC 4889822. PMID 27298622.
- Perdigão, Nelson; et al. (2015). "Unexpected features of the dark proteome". PNAS. 112 (52): 15898–15903. Bibcode:2015PNAS..11215898P. doi:10.1073/pnas.1508380112. PMC 4702990. PMID 26578815.
- Adhikari, S (October 2020). "A high-stringency blueprint of the human proteome". Nature Communications. 11 (1): 5301. doi:10.1038/s41467-020-19045-9. PMC 7568584. PMID 33067450.
- Altelaar, AF; Munoz, J; Heck, AJ (January 2013). "Next-generation proteomics: towards an integrative view of proteome dynamics". Nature Reviews Genetics. 14 (1): 35–48. doi:10.1038/nrg3356. PMID 23207911. S2CID 10248311.
- Wilhelm, Mathias; Schlegl, Judith; Hahne, Hannes; Gholami, Amin Moghaddas; Lieberenz, Marcus; Savitski, Mikhail M.; Ziegler, Emanuel; Butzmann, Lars; Gessulat, Siegfried; Marx, Harald; Mathieson, Toby; Lemeer, Simone; Schnatbaum, Karsten; Reimer, Ulf; Wenschuh, Holger; Mollenhauer, Martin; Slotta-Huspenina, Julia; Boese, Joos-Hendrik; Bantscheff, Marcus; Gerstmair, Anja; Faerber, Franz; Kuster, Bernhard (2014). "Mass-Spectrometry-Based Draft of the Human Proteome". Nature. 509 (7502): 582–7. Bibcode:2014Natur.509..582W. doi:10.1038/nature13319. PMID 24870543. S2CID 4467721.
- Kim, Min-Sik; et al. (May 2014). "A draft map of the human proteome". Nature. 509 (7502): 575–81. Bibcode:2014Natur.509..575K. doi:10.1038/nature13302. PMC 4403737. PMID 24870542.
- Shi, Yang; Xiang, Rong; Horváth, Csaba; Wilkins, James A. (2004-10-22). "The role of liquid chromatography in proteomics". Journal of Chromatography A. Bioanalytical Chemistry: Perspectives and Recent Advances with Recognition of Barry L. Karger. 1053 (1): 27–36. doi:10.1016/j.chroma.2004.07.044. ISSN 0021-9673. PMID 15543969.
- "Pull-Down Assays - US". www.thermofisher.com. Retrieved 2019-12-05.
External links
- PIR database
- UniProt database
- Pfam database at the Library of Congress Web Archives (archived 2011-05-06)