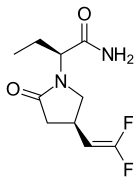
Seletracetam
Seletracetam (UCB 44212) is a pyrrolidone-derived[2] drug of the racetam family that is structurally related to levetiracetam (trade name Keppra).[2][3] It was under development by UCB Pharmaceuticals as a more potent and effective anticonvulsant drug to replace levetiracetam but its development has been halted.[1]
 | |
| Clinical data | |
|---|---|
| Routes of administration | Oral[1][2] |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | >90%[1][2] |
| Elimination half-life | 8 hours[1][2] |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C10H14F2N2O |
| Molar mass | 232.227 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
There are two main mechanisms of action for seletracetam. The first is its high-affinity stereospecific binding to synaptic vesicle glycoprotein 2A (SV2A).[2][3] Seletracetam has shown potent seizure suppression in models of acquired and genetic epilepsy,[3] and has been well tolerated by various animal models.[3] The second is its binding to N-type calcium channels and preventing influx of Ca2+ during high-voltage activation that is typical of epilepsy.[4][5][6]
While similar in structure to nootropic drugs, it is not expected to have cognitive enhancing properties.[7] Seletracetam was in Phase II clinical trials under the supervision of the U.S. Food and Drug Administration (FDA) but its production is on hold.[3]
Synthesis
Seletracetam's molecular structure contains elements common to other anticonvulsants, including levetiracetam and brivaracetam, such as a nitrogen heterocyclic system.[8][9] Like brivaracetam, seletracetam is a derivative of levetiracetam.
Structure and activity relationship studies have concluded that the most potent anticonvulsant activity was at the imide nitrogen atom and that this activity was further enhanced by nearby electronegative functional groups such as the di-fluoro group on seletracetam.[10]
Administration
Seletracetam is an orally administered drug, after which it is quickly and efficiently absorbed.[2] The typical dosage is 0.03–10 mg/kg per day (up to 0.6g per day).[1]
Mechanism of action
Seletracetam's anti-epileptic effects are due to its high affinity binding to synaptic vesicle glycoprotein 2A (SV2A)[1][2][3]—part of a calcium ion regulator. The SV2A protein assists with the coordination of synaptic vesicle exocytosis,[11][12] which induces neurotransmitter release in the presence of an influx in Ca2+. A correlation has been drawn between the binding affinity of seletracetam (and its analogues) to SV2A and the degree of seizure prevention in animal models.[7][11][13]
In addition, studies of ion currents have shown that seletracetam significantly decreases the amount of high-voltage derived Ca2+ currents[4] which have been implicated in causing the high intracellular Ca2+ influx during epileptic activity.[5] It is thought that seletracetam binds to N-type Ca2+ channels and inhibits their ability to allow calcium ions to enter the cell,[4][5] although the drug does not bind to T-type channels that mediate low-voltage activated Ca2+ currents.[2][14] Seletracetam thereby decreases cellular excitation, but it does not seem to affect voltage-gated Na+ or K+ currents.[4] Selectracetam has been demonstrated to not significantly affect currents gated by NMDA, AMPA, GABA, glycine, or kainic acid.[15]
The dual effect of seletracetam is an overall decrease in the amount of Ca2+ influx in the cell during an action potential due to binding at N-type channels, which prevents over-excitation of the neuron, as well as a decrease in neurotransmitter release as a product of cellular excitation due to the interaction of the drug with SV2A, which reduces the spread of excitation to nearby cells.[6]
Compared to levetiracetam, which binds at the same site,[11] seletracetam binds to SV2A with ten times higher affinity.[7][13]
The nature of why the seletracetam molecule binds so specifically to SV2A and how SV2A affects exocytosis is unclear.[16]
Pharmacodynamics and pharmacokinetics
The oral bioavailability of seletracetam is >90%[2] and its half-life is approximately 8 hours.[1][2] 25% of ingested seletracetam is metabolized and excreted unchanged and about 53% is excreted in the form of an inactive carboxylic acid metabolite.[11][14] The main metabolic mechanism is the hydrolysis of an acetamide to a carboxylic acid.[14][17]
Seletracetam exhibits first-order mono-compartmental pharmacokinetics, in which there is a simple linear relationship between the amount of drug that was administered, the time that has passed, and the amount of drug subsequently remaining in the body.[2] This contrasts the nonlinear pharmacokinetics typical of previously available anticonvulsants such as phenobarbital, phenytolin, valproate and carbamazepine.[2] The benefit of linear kinetics is that the steady-state concentration of the drug is directly and reliably related to the dose of the drug that is administered; this allows for simple and reliable dose adjustments.
In vitro studies
In vitro studies performed in rodent hippocampal slices found that seletracetam causes a complete reversal of the increases in activity of population spike amplitude in epilepsy models.[7] These reductions in in vitro epilepsy symptoms were present at extracellular concentrations of 3.2 μM.[7] This is approximately 10% of the most effective concentration of levetiracetam in similar tests.[18]
Animal studies
Seletracetam has been tested on various animal models for epilepsy, with mixed results.
Unlike drugs that act on voltage-gated sodium channels,[19] seletracetam was demonstrated to have no significant effect on the maximal electroshock seizure test results in mice.[7] It similarly had no relieving effects in mice of the other most common acute seizure model, the pentylenetetrazol convulsion-induction model.[2][7][14]
Seletracetam did, however, show promising results in acquired and genetic epilepsy models.[2][7] In the mouse model of corneal kindling, which exhibits the anticonvulsant capability of generalized motor seizures, doses as low as 0.07 mg/kg intraperitoneal injection (i.p.), and ED50 of 0.31 mg/kg i.p. were effective.[7] Occurrence of audiogenic seizures—those induced by white noise—in mice were also significantly reduced by an ED50 of 0.17 mg/kg i.p., which suggests that selectracetam reduces convulsions caused by clonic seizures.[7]
In hippocampal kindling model rats, seizure severity was significantly reduced by seletracetam oral doses of 0.23 mg/kg. This provides further evidence of the potential benefits of selectracetam on generalized motor seizures.[7] Seletracetam also performed well as a method to reduce the suppression of spontaneous spike-and-wave discharges that are often associated with absence epilepsy activity.[20] This was demonstrated by its effect on Strasbourg genetic absence epilepsy rats (GAERS).[21] This model had an ED50 of 0.15 mg/kg i.p.[7]
Rodents were found to have negligible behavioral deficits as a result of seletracetam administration, as measured by performance on a rotarod task.[7][22]
Adverse effects and tolerance
Unlike currently prescribed anticonvulsants such as phenytoin, valproate, and phenobarbital, seletracetam shows few central nervous system (CNS) side effects and is predicted to have low levels of drug-drug interactions due to its low binding (<10%) to plasma proteins.[2][23] There have been, however, no formal studies conducted on drug-drug interactions with seletracetam.[11]
Other than SV2A and the high-voltage-activated Ca2+ channels, seletracetam does not significantly bind to other CNS receptors, ion channels, or uptake mechanisms.[11] Seletracetam has, however, shown a slight selectivity for glycine receptors.[11][14][24] This drug neither inhibits nor unnecessarily induces the action of any major human metabolizing enzymes, which further reduces adverse effects.[23]
Early data from phase I trials were optimistic, and found seletracetam to be well tolerated by human volunteers.[25]
In phase II trials side effects were limited to the CNS in origin, were of mild to moderate severity, and most were resolved within 24 hours[2] and with no medical intervention.[11] The most frequently reported adverse effects of seletracetam were dizziness, feeling drunk, euphoria, nausea, and somnolence.[11]
Seletracetam was well tolerated by healthy individuals after single oral doses ranging from 2 to 600 mg, as well as after b.i.d. (twice daily) doses of 200 mg.[11] Toxicology studies have shown that this drug has low acute oral toxicity and no significant negative effects on the CNS, cardiac, or respiratory systems.[11] High doses of 2000 mg/kg per day (in mice and rats) and greater than 600 mg/kg per day (in dogs) were poorly tolerated.[11]
FDA approval status
Phase II clinical trials of seletracetam were ongoing but in July 2007 the company stated that the drug's development has been put on hold.[3] Although the conducted Phase II trials showed success, it was less than expected given the performance of seletracetam in animal models.[1] There have been no known Phase IIb or Phase III trials.[3]
In 2010, development of seletracetam was halted in favor of the development of brivaracetam, a newer variation of the drug.[1]
References
- Malykh, AG; Sadaie, MR (Feb 12, 2010). "Piracetam and piracetam-like drugs: from basic science to novel clinical applications to CNS disorders". Drugs. 70 (3): 287–312. doi:10.2165/11319230-000000000-00000. PMID 20166767. S2CID 12176745.
- Bennett, B; Matagne, A; Michel, P; Leonard, M; Cornet, M; Meeus, MA; Toublanc, N (January 2007). "Seletracetam (UCB 44212)". Neurotherapeutics. 4 (1): 117–22. doi:10.1016/j.nurt.2006.11.014. PMC 7479702. PMID 17199025.
- Pollard, JR; Kulig, K (January 2008). "Seletracetam, a small molecule SV2A modulator for the treatment of epilepsy". Current Opinion in Investigational Drugs. 9 (1): 101–7. PMID 18183537.
- Martella, G; Bonsi, P; Sciamanna, G; Platania, P; Madeo, G; Tassone, A; Cuomo, D; Pisani, A (April 2009). "Seletracetam (ucb 44212) inhibits high-voltage-activated Ca2+ currents and intracellular Ca2+ increase in rat cortical neurons in vitro". Epilepsia. 50 (4): 702–10. doi:10.1111/j.1528-1167.2008.01915.x. PMID 19055493. S2CID 9067249.
- Pisani A, Bonsi P, Martella G, De Persis C, Costa C, Pisani F, Bernardi G, Calabresi P (July 2004). "Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine". Epilepsia. 45 (7): 719–28. doi:10.1111/j.0013-9580.2004.02204.x. PMID 15230693. S2CID 31781002.
- Custer, KL; Austin, NS; Sullivan, JM; Bajjalieh, SM (Jan 25, 2006). "Synaptic vesicle protein 2 enhances release probability at quiescent synapses". The Journal of Neuroscience. 26 (4): 1303–13. doi:10.1523/JNEUROSCI.2699-05.2006. PMC 6674579. PMID 16436618.
- Matagne, A; Margineanu, DG; Potschka, H; Löscher, W; Michel, P; Kenda, B; Klitgaard, H (Jul 1, 2009). "Profile of the new pyrrolidone derivative seletracetam (ucb 44212) in animal models of epilepsy". European Journal of Pharmacology. 614 (1–3): 30–7. doi:10.1016/j.ejphar.2009.04.024. PMID 19383493.
- Wong, MG; Defina, JA; Andrews, PR (April 1986). "Conformational analysis of clinically active anticonvulsant drugs". Journal of Medicinal Chemistry. 29 (4): 562–72. doi:10.1021/jm00154a022. PMID 3959032.
- Bruno-Blanch, L; Gálvez, J; García-Domenech, R (Aug 18, 2003). "Topological virtual screening: a way to find new anticonvulsant drugs from chemical diversity". Bioorganic & Medicinal Chemistry Letters. 13 (16): 2749–54. doi:10.1016/S0960-894X(03)00535-3. PMID 12873507.
- Kamiński, K; Rzepka, S; Obniska, J (Oct 1, 2011). "Synthesis and anticonvulsant activity of new 1-[2-oxo-2-(4-phenylpiperazin-1-yl)ethyl]pyrrolidine-2,5-diones". Bioorganic & Medicinal Chemistry Letters. 21 (19): 5800–3. doi:10.1016/j.bmcl.2011.07.118. PMID 21875804.
- Bialer, M; Johannessen, SI; Kupferberg, HJ; Levy, RH; Perucca, E; Tomson, T (January 2007). "Progress report on new antiepileptic drugs: a summary of the Eighth Eilat Conference (EILAT VIII)". Epilepsy Research. 73 (1): 1–52. doi:10.1016/j.eplepsyres.2006.10.008. PMID 17158031. S2CID 45026113.
- Crowder, KM; Gunther, JM; Jones, TA; Hale, BD; Zhang, HZ; Peterson, MR; Scheller, RH; Chavkin, C; Bajjalieh, SM (Dec 21, 1999). "Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A)". Proceedings of the National Academy of Sciences of the United States of America. 96 (26): 15268–73. doi:10.1073/pnas.96.26.15268. PMC 24809. PMID 10611374.
- Lynch, BA; Lambeng, N; Nocka, K; Kensel-Hammes, P; Bajjalieh, SM; Matagne, A; Fuks, B (Jun 29, 2004). "The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam". Proceedings of the National Academy of Sciences of the United States of America. 101 (26): 9861–6. doi:10.1073/pnas.0308208101. PMC 470764. PMID 15210974.
- Luszczki, JJ (Mar–Apr 2009). "Third-generation antiepileptic drugs: mechanisms of action, pharmacokinetics and interactions". Pharmacological Reports : PR. 61 (2): 197–216. doi:10.1016/s1734-1140(09)70024-6. PMID 19443931.
- Rigo, J.M.; L. Nguyen; G. Hans; S. Belachew; G. Moonen; A. Matagne; H. Klitgaard (2005). "Seletracetam (ucb 44212): effect on inhibitory and excitatory neurotransmission". Epilepsia. 46 (Suppl. 8): 110. doi:10.1111/j.1528-1167.2005.460801_14.x. S2CID 221732819.
- Pollard, John R; French, Jacqueline (1 December 2006). "Antiepileptic drugs in development". The Lancet Neurology. 5 (12): 1064–1067. doi:10.1016/S1474-4422(06)70627-5. PMID 17110287. S2CID 22569327.
- Strolin Benedetti, M; Whomsley, R; Nicolas, JM; Young, C; Baltes, E (November 2003). "Pharmacokinetics and metabolism of 14C-levetiracetam, a new antiepileptic agent, in healthy volunteers". European Journal of Clinical Pharmacology. 59 (8–9): 621–30. doi:10.1007/s00228-003-0655-6. PMID 14530892. S2CID 25017909.
- Margineanu, DG; Klitgaard, H (October 2000). "Inhibition of neuronal hypersynchrony in vitro differentiates levetiracetam from classical antiepileptic drugs". Pharmacological Research. 42 (4): 281–5. doi:10.1006/phrs.2000.0689. PMID 10987984.
- Rogawski MA (June 2006). "Diverse mechanisms of antiepileptic drugs in the development pipeline". Epilepsy Res. 69 (3): 273–94. doi:10.1016/j.eplepsyres.2006.02.004. PMC 1562526. PMID 16621450.
- Hughes, JR; Cortés Mdel, C (August 2009). "Absence seizures: a review of recent reports with new concepts". Epilepsy & Behavior. 15 (4): 404–12. doi:10.1016/j.yebeh.2009.06.007. PMID 19632158. S2CID 22023692.
- Marescaux, C; Vergnes, M; Depaulis, A (1992). Genetic absence epilepsy in rats from Strasbourg--a review. Journal of Neural Transmission. Supplementum. 35. pp. 37–69. doi:10.1007/978-3-7091-9206-1_4. ISBN 978-3-211-82340-8. PMID 1512594.
- Stefan, H; Steinhoff, BJ (October 2007). "Emerging drugs for epilepsy and other treatment options". European Journal of Neurology. 14 (10): 1154–61. doi:10.1111/j.1468-1331.2007.01706.x. PMID 17880570. S2CID 41155699.
- Brodie, MJ (May 2001). "Do we need any more new antiepileptic drugs?". Epilepsy Research. 45 (1–3): 3–6. doi:10.1016/S0920-1211(01)00203-0. PMID 11461782. S2CID 37908949.
- Bialer, M; Johannessen, SI; Kupferberg, HJ; Levy, RH; Perucca, E; Tomson, T (Sep–Oct 2004). "Progress report on new antiepileptic drugs: a summary of the Seventh Eilat Conference (EILAT VII)". Epilepsy Research. 61 (1–3): 1–48. doi:10.1016/j.eplepsyres.2004.07.010. PMID 15570674. S2CID 1154454.
- Leese, P.T.; D.R. Goldwater; R. Hulhoven; E. Salas; N. Toublanc; D. Chen; M.L. Sargentini-Maier; A. Stockis (2006). "Seletracetam (UCB 44212): Single and Multiple Rising Dose Safety, Tolerability and Pharmacokinetics in Healthy Subjects". American Epilepsy Society Abstracts: 2.131.