Histidinemia
Histidinemia, is a rare autosomal recessive metabolic disorder caused by a deficiency of the enzyme histidase. Histidase is needed for the metabolism of the amino acid histidine.[1] Although originally thought to be linked to multiple developmental disorders histidinemia is now accepted as a relatively benign disorder,[2] leading to a reduction in the prevalence of neonatal screening procedures.[3]
| Histidinemia | |
|---|---|
| Other names | Histidinuria |
 | |
| Histidine | |
| Specialty | Endocrinology |
Presentation
Histidinemia is considered benign as most patients remain asymptomatic, early correlational evidence from the first decade of histidinemia research lead to the theory that histidinemia was associated with multiple developmental symptoms including hyperactivity, speech impediment, developmental delay, learning difficulties, and sometimes mental retardation.[3] However, these claims were later deemed coincidental as a large subpopulation of infants that tested positive for histidinemia were found to have normal IQ and speech characteristics; as such histidinemia has since been reclassified as a benign inborn error of metabolism.[2]
Molecular mechanism
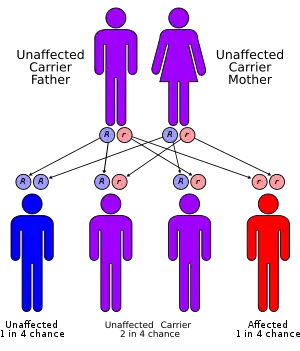
Histidinemia occurs as the result of an inborn error of metabolism that may result in either an inactive or a severely reduced Histidine ammonia-lyase enzyme activity. The gene that encodes for Histidine ammonia-lyase (HAL) spans a roughly 25 kb and consists of 21 exons[4] located at the 12q22-q24.1 position of human chromosome 12.[5] There are 8 mutations currently associated with autosomal recessive histidinemia, that include: four missense mutations, two exonic polymorphisms and two intronic polymorphisms.[5]
Diagnosis
Histidenemia is characterized by increased levels of histidine, histamine and imidazole in blood, urine and cerebrospinal fluid. This also results in decreased levels of the metabolite urocanic acid in blood, urine, and skin cells.[1] In Japan, neonatal screening was previously performed on infants within 1 month of birth; infants demonstrating a blood histidine level of 6 mg/dl or more underwent careful testing as suspected histidinemia cases.[6] A typical characteristic of histidinemia is an increase in the blood histidine levels from normal levels (70-120 μM) to an elevated level (290-1420 μM).[3] Further testing includes: observing histidine as well as imidazolepyruvic acid metabolites in the urine. However, neonatal urine testing has been discontinued in most places, with the exception of Quebec.[3]
Treatment
It has been suggested that a possible method of treatment for histidinemia is through the adoption of a diet that is low in histidine intake. However, the requirement for such dietary restrictions is typically unnecessary for 99% of all cases of histidinemia.[3]
Prevalence
Histidinemia is a rare autosomal recessive disorder. However, histidinemia is considered the most prevalent inborn error of metabolism[3][5] with a reported incidence of 1:8600 (Quebec); 1:180,000 (New York) and 1:9600 (Japan); and an average of 1:12,000 observed in the neonatal screening of over 20 million newborns.[3]
See also
References
- Taylor RG, Levi HL, McInnes RR (1991). "Histidase and histidinemia. Clinical and molecular considerations". Molecular Biology & Medicine. 8 (1): 101–116. PMID 1943682.
- Lam WK, Cleary MA, Wraith JE, Walter JH (1996). "Histidinemia: a benign metabolic disorder". Archives of Disease in Childhood. 74 (4): 343–346. doi:10.1136/adc.74.4.343. PMC 1511463. PMID 8669938.
- Levy H (2004). "Histidinemia" (PDF). Orphanet.
- Suchi M, Sano H, Mizuno H, Wada Y (1995). "Molecular cloning and structural characterization of the human histidase gene (HAL)". Genomics. 29 (1): 98–104. doi:10.1006/geno.1995.1219. PMID 8530107.
- Kawai Y, Moriyama A, Asai K, Coleman-Campbell CM, Morishita H, Suchi M (2005). "Molecular characterization of histidinemia: identification of four missense mutations in the histidase gene". Human Genetics. 116 (5): 340–346. doi:10.1007/s00439-004-1232-5. PMID 15806399. S2CID 33960184.
- Kitagawa T, Owada M, Sakiyama T, Kojima T, Kondo T (1981). "Experience and problems of newborn mass screening for inborn errors of metabolism in Japan". Acta Paediatrica Japonica. 23 (1): 24–34. doi:10.1111/j.1442-200x.1981.tb01242.x.
External links
| Classification |
|---|