Tyrosinemia type III
Tyrosinemia type III is a rare disorder caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase (EC 1.13.11.27), encoded by the gene HPD.[2] This enzyme is abundant in the liver, and smaller amounts are found in the kidneys. It is one of a series of enzymes needed to break down tyrosine. Specifically, 4-hydroxyphenylpyruvate dioxygenase converts a tyrosine byproduct called 4-hydroxyphenylpyruvate to homogentisic acid. Characteristic features of type III tyrosinemia include mild mental retardation, seizures, and periodic loss of balance and coordination (intermittent ataxia). Type III tyrosinemia is very rare; only a few cases have been reported.[2]
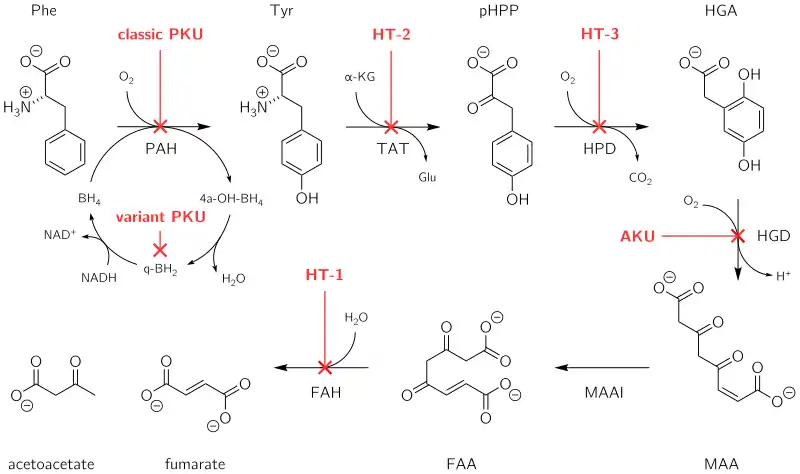
Pathophysiology of metabolic disorders of tyrosine, resulting in elevated levels of tyrosine in blood.
| Tyrosinemia type III | |
|---|---|
| Other names | TYRSN3[1] |
 | |
| Tyrosine | |
| Specialty | Endocrinology |
References
- "OMIM Entry - # 276710 - TYROSINEMIA, TYPE III; TYRSN3". omim.org.
- Zea-Rey AV, Cruz-Camino H, Vazquez-Cantu DL, Gutiérrez-García VM, Santos-Guzmán J, Cantú-Reyna C (27 November 2017). "The Incidence of Transient Neonatal Tyrosinemia Within a Mexican Population" (PDF). Journal of Inborn Errors of Metabolism and Screening. 5: 232640981774423. doi:10.1177/2326409817744230.
External links
| Classification | |
|---|---|
| External resources |
This article is issued from Wikipedia. The text is licensed under Creative Commons - Attribution - Sharealike. Additional terms may apply for the media files.