Tyrosinemia type I
Tyrosinemia type I is a genetic disorder that disrupts the metabolism of the amino acid tyrosine, resulting in damage primarily to the liver along with the kidneys and peripheral nerves.[1] The inability of cells to process tyrosine can lead to chronic liver damage ending in liver failure, as well as renal disease and rickets. Symptoms such as poor growth and enlarged liver are associated with the clinical presentation of the disease.[2] Clinical manifestation of disease occurs typically within the first two years of life. The severity of the disease is correlated with the timing of onset of symptoms, earlier being more severe.[1]
| Tyrosinemia type I | |
|---|---|
| Other names | Hereditary Tyrosinemia type I, HT1 |
 | |
| Mutation of enzyme fumarylacetoacetate hydrolase (FAH) in the tyrosine catabolic pathway | |
| Specialty | Hepatology, nephrology, neurology |
| Symptoms | Failure to thrive, enlarged liver, fever, vomiting, diarrhea |
| Usual onset | Variable, usually with the first 2 years of life |
| Duration | Lifelong |
| Causes | Genetic (autosomal recessive) |
| Diagnostic method | Dried blood spot testing, urinalysis, genetic testing |
| Treatment | Dietary restrictions, Nitisinone, liver transplantation |
| Medication | Nitisinone |
| Prognosis | 93% survival rate at six years with treatment |
| Frequency | 1 in 1,850 (Saguenay-Lac Saint-Jean region, Quebec) |
Tyrosinemia type I is an autosomal recessive disorder caused by mutations in the both copies of the gene encoding the enzyme fumarylacetoacetate hydrolase (FAH). FAH is a metabolic enzyme that catalyzes the conversion of fumarylacetoacetate to fumarate and acetoacetate. It is expressed primarily in the liver and kidney. Loss of FAH activity results in the accumulation of certain metabolic intermediates in the tyrosine catabolic pathway.[2] These compounds are toxic to cells and lead to differential gene expression and apoptosis in high concentrations.[2] HT1 is diagnosed when elevated levels of succinylacetone (SA), one of the metabolites in this pathway, is detected in blood and urine samples.[1]
While there is no cure for tyrosinemia type I, management of the disease is possible utilizing dietary restrictions and medications. A diet low in tyrosine and phenylalanine is utilized indefinitely once a diagnosis is suspected or confirmed. Additionally, the drug nitisinone (brand name Orfadin) is prescribed and continued indefinitely in order to combat liver and kidney damage, promoting normal function of these organs.[1] Prior to the development of nitisinone, dietary restrictions and liver transplantation were the only forms of treatment for HT1.[2]
Tyrosinemia type I is especially prevalent in the Saguenay-Lac Saint-Jean region of Quebec, where the prevalence is 1 in 1,850 births. It is most common among those with French-Canadian ancestry and this frequency of infliction has been attributed to the founder effect.[3] There are five other known types of tyrosinemia, all of which derange the metabolism of tyrosine in the human body. They are distinguished by their symptoms and genetic cause.[2]
Signs and symptoms
Type 1 tyrosinemia typically presents in infancy as failure to thrive and hepatomegaly. The primary effects are progressive liver and kidney dysfunction. The liver disease causes cirrhosis, conjugated hyperbilirubinemia, elevated AFP, hypoglycemia and coagulation abnormalities. This can lead to jaundice, ascites and hemorrhage. There is also an increased risk of hepatocellular carcinoma.The kidney dysfunction presents as Fanconi syndrome: Renal tubular acidosis, hypophosphatemia and aminoaciduria. Cardiomyopathy, neurologic and dermatologic manifestations are also possible. The urine has an odor of cabbage or rancid butter.[4]
The presentation of symptoms of tyrosinemia type 1 in terms of timing is broken into three categories: acute, sub-acute, and chronic.
The acute classification typically is presented clinically between birth and 6 months of age. The common presentation in an acute case is synthetic liver failure, marked by the lack of formation of coagulation factors in blood. Patients are prone to infections at this stage accompanied by fever, vomiting, increased tendency to bleed, and diarrhea along with bloody feces as manifestations of sepsis. Other symptoms include enlarged liver, jaundice, and excess abdominal fluid.[1][2]
Sub-acute cases present between 6 months and the first year of life and the severity of liver disease is lessened to an extent. Again, synthetic function of the liver in terms of blood coagulation factors is impaired in addition to enlargement of the liver and spleen. The infant may also display a failure to thrive as their growth is limited by the disease. This growth impairment can manifest itself in rickets, which is the softening of bones.[1][2]
The final classification, chronic HT1, is detected with presentations occurring after one year of life. The course of the disease up to this point can lead to different ailments affecting the liver. Cirrhosis, liver failure, or cancer of the liver may present as a result of chronic liver disease. Additional symptoms common in this classification include cardiomyopathy, renal disease, and acute neurological crises.[1][2]
Liver
The liver is the organ affected most by Tyrosinemia Type I due to the high level of expression of the gene for fumarylacetoacetate hydrolase (FAH) in liver cells. The production of blood coagulation factors by the liver is disrupted, causing hemophiliac-like symptoms. Acute liver failure is common, especially in early life. Additionally, the synthesis of albumin in the liver may be defective, therefore leading to hypoalbuminemia. As the disease progresses, cirrhosis is common. This can lead to a fatty liver and the development of tumors in areas affected by this scarring of liver tissue. These scars are known as nodules.[2] There is a 37% chance of developing a hepatocellular carcinoma (HCC) for untreated patients.[5]
Renal and neurological manifestations
Many patients display impaired kidney function and neurological symptoms. In addition to liver cells, kidney cell expression involves expression of the gene for FAH. Kidney failure is a potential result of impaired kidney function, but the most common symptom associated with renal dysfunction is hypophosphatemic rickets.[2] Neurological manifestations are characterized by acute neurological crises due to overaccumulation of porphyrin. These crises are characterized by porphyria. They typically follow an infection. Patients can present with a variety of varied symptoms including paresthesias, abdominal pain, pain-induced seizures, and can result in self-mutilation in response to this pain. Episodes can last for 1–7 days and can lead to neuropathy.[1][2]
Genetics
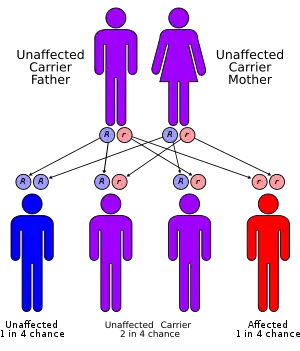
Tyrosinemia type I is an autosomal recessive inherited condition. Mutant alleles in the gene are inherited from both parents. The genetic mutation occurs to the fumarylacetoacetate hydrolase (FAH) enzyme gene, located on chromosome 15. The most common mutation is IVS12+5(G->A) which is a mutation in the splice site consensus sequence of intron 12, therefore affecting exon 12. A second allele is the IVS6-1(G-T) mutation. This mutation results in a nonfunctional enzyme.[2]
Type 1 tyrosinemia is inherited in an autosomal recessive pattern.[6]
Pathophysiology
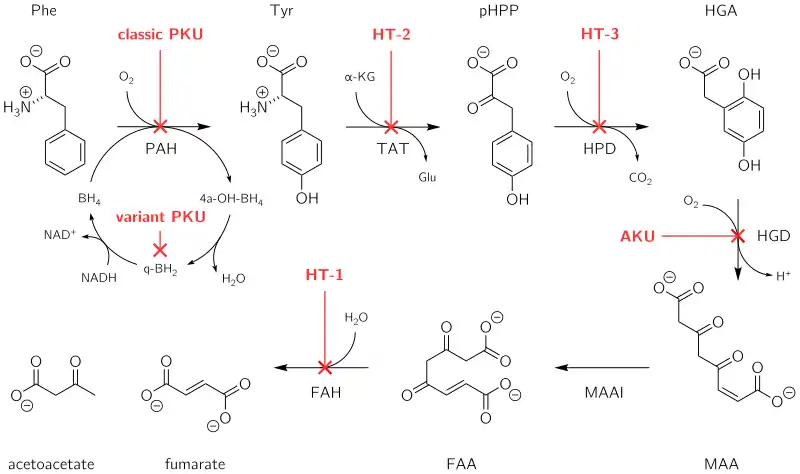
Fumarylacetoacetate hydrolase catalyzes the final step in the degradation of tyrosine - fumarylacetoacetate to fumarate, acetoacetate and succinate. Fumarylacetoacetate accumulates in hepatocytes and proximal renal tubal cells and causes oxidative damage and DNA damage leading to cell death and dysfunctional gene expression which alters metabolic processes like protein synthesis and gluconeogenesis. The increase in fumarylacetoacetate inhibits previous steps in tyrosine degradation leading to an accumulation of tyrosine in the body. Tyrosine is not directly toxic to the liver or kidneys but causes dermatologic and neurodevelopmental problems.
Tyrosine metabolic pathway
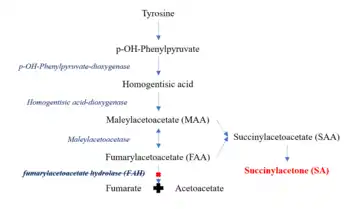
Fumarylacetoacetate hydrolase (FAH) is the final enzyme in the tyrosine metabolic pathway.[1] The mutation of FAH enzyme results in nonfunctional FAH in all cells expressing this gene and thus metabolizing tyrosine is impaired. FAH catalyzes the conversion of fumarylacetoacetate to fumarate and acetoacetate. Loss of FAH results in the accumulation of upstream compounds in the catabolic pathway. These include maleylacetoacetate (MAA) and fumarylacetoacetate (FAA). MAA and FAA are converted to succinylacetoacetate (SAA) which is then catabolized to succinylacetone (SA).[2]
The accumulation of MAA, FAA, and SA in cells inhibits the breakdown of thiol derivatives, leading to post-translational modifications to the antioxidant glutathione. This inhibits the antioxidant activity of glutathione, leading to reactive oxygen species (ROS) damaging cell components. Over time, the combined effect of accumulation of toxic metabolic intermediates and elevated ROS levels in liver and kidney cells leads to apoptosis in these tissues which ultimately results in organ failure.[2] Accumulated SA in liver and kidney cells results in its release into the bloodstream, which leads to secondary effects. SA inhibits the enzyme 5-ALA dehydratase which converts aminolevulinic acid (5-ALA) into porphobilinogen, a precursor to porphyrin. Consequently, porphyrin deposits form in the bloodstream and cause neuropathic pain, leading to the acute neurological crises experienced by some patients. Additionally. SA can function to inhibit renal tubular function, the synthesis of heme, and the immune system.[2]
The accumulation of unprocessed tyrosine itself in the blood stream as a consequence of deficient catabolism can also lead to disruption of hormonal signaling and neurotransmission. Tyrosine is a precursor molecule required for synthesis of several neurotransmitters and hormones, mainly Dopamine, norepinephrine, and thryoxine. Excessive synthesis of these molecules due to elevated tyrosine levels can impair physical growth, motor function, and speech development.[7][8]
Diagnosis
Beyond the identification of physical clinical symptoms outlined above, the definitive criterion for diagnostic assessment of Tyrosinemia Type I is elevated succinylacetone (SA) in blood and urine. Elevated SA levels are not associated with any other known medical condition, so there is minimal risk of misdiagnosis.[5] Quantitation of tyrosine levels is also used as a diagnostic but is less reliable due to high false positive and false negative rates.[9] Newborns are not generally screened for HT1 due to rarity of the condition and lack of apparent symptoms at time of birth.[1] However, prompt assessment upon the manifestation of physical symptoms such as fever, vomiting, increased tendency to bleed, diarrhea along with bloody feces, and jaundice is critical for improving long term prognosis.[1][2]
Management
The primary treatment for type 1 tyrosinemia is nitisinone and restriction of tyrosine in the diet.[6] Nitisinone inhibits the conversion of 4-OH phenylpyruvate to homogentisic acid by 4-Hydroxyphenylpyruvate dioxygenase, the second step in tyrosine degradation. By inhibiting this enzyme, the accumulation of the fumarylacetoacetate is prevented.[10] Previously, liver transplantation was the primary treatment option and is still used in patients in whom nitisinone fails.
Clinical treatment of HT1 relies on medications and strict regulation of diet. Nitisinone and dietary restrictions that decrease the amount of tyrosine and phenylalaine absorbed from the GI tract during protein digestion are used in combination as therapeutic measures that control the disease state if they are continued indefinitely. If not, there is a lack of control over the disease, resulting in continued liver and kidney damage, contributing to organ failure and death. In this case, a liver transplant may be required.[2] Levels of SA are monitored throughout treatment in order to assess treatment effectiveness.[9]
Diet
The prescribed diet for treatment of HT1 is low in protein. Patients received amino acid supplements lacking tyrosine and phenylalanine in order to acquire sufficient protein. It is recommended that tyrosine levels remain below 500 μmol/L.[5] Phenylalnine is the precursor to tyrosine. The ideology behind maintaining low tyrosine levels is two-fold. Firstly, it prevents the toxic metabolic intermediates from accumulating as a result of the dysfunctional tyrosine metabolic pathway. Prior to the introduction of nitisinone, this was the main treatment measure. Secondly, the mechanism of action of nitisinone is prevention of any tyrosine metabolism, thus it is important to prevent tyrosine from accumulating. Dietary protein consumption while taking nitisinone can also lead to side effects affecting the ocular system, which are easily reversed by removing protein from the diet.[9]
Medication
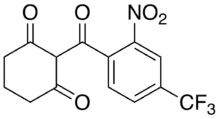
Nitisinone is prescribed ultimately to reduce the accumulation of toxic metabolic intermediates, such as succinylacetate, which are toxic to cells. It modifies the function of 4-hydrooxyphenylpyruvate dioxygenase by acting as a competitive inhibitor. 4-hydrooxyphenylpyruvate dioxygenase functions to convert 4-hydroxyphenylpyruvate to homogentisate as the second enzymatic reaction in the tyrosine catabolic pathway. This prevents the further catabolism of tyrosine.[5] It is recommended that nitisinone treatment begins immediately following a confirmed or suspected case of HT1.[1] It is supplied orally as a capsule or suspension in dose increments of 2 mg, 5 mg, 10 mg, or 20 mg or 4 mg/mL respectively.[5] The starting dose is 1 mg/kg one time daily or 2 mg/kg one time daily for 48 hours if the patient is experiencing acute liver failure. Patient responsiveness to nitisinone is assessed by measuring blood coagulation activity and SA levels in blood and urine. Patients should display a positive response within 24–48 hours of first dose. Establishment of the long-term dosage will vary from patient to patient. It is recommended that nitisinone levels be maintained at 30-50 μM in the blood stream.[1]
Prognosis
Prior to the development of nitisinone, dietary restrictions and liver transplantation were the only forms of treatment for HT1.[2] A study regarding the efficacy of treatment with nitisinone and dietary restrictions found that 93% of people survived at two years, four years, and six years indicating the prognosis of stabilizing the HT1 disease state is positive.[5]
Epidemiology
Tyrosinemia type I affects males and females in equal numbers. Its prevalence has been estimated to be 1 in 100,000 to 120,000 births worldwide. HT1 is especially prevalent in the Saguenay-Lac Saint-Jean region of Quebec is one in 1,850 births. The elevated frequency of this disorder within individuals of French-Canadian ancestry in Quebec is believed to be due to reduced genetic heterogeneity within the original founder population for the Saguenay-Lac Saint-Jean region.[11] The initial settlement of Saguenay Lac-Saint-Jean (SLSJ) occurred between 1838 and 1911. From a total of 28,656 settlers, 75 percent originated from the neighboring Charlevoix region. The settling of the Charlevoix region itself started in 1675 when 599 founders of mostly French descent moved to this region from the Quebec City area.
Worldwide, type I tyrosinemia affects about 1 person in 100,000. This type of tyrosinemia is much more common in Quebec, Canada. The overall incidence in Quebec is about 1 in 16,000 individuals. In the Saguenay-Lac-Saint-Jean region of Quebec, type 1 tyrosinemia affects 1 person in 1,846.[12] The carrier rate has been estimated to be between 1 in 20 and 1 in 31.[13]
History
Nitisinone was first used to clinically treat tyrosinemia type I in 1991. Nitisinone was approved by the European Medicine Agency (EMA) under exceptional circumstances in 2005. Originally, nitisinone was developed as a weed-killer by Zeneca Agrochemicals. It was epidemiologically observed that the growth of plants and weeds was inhibited under the bottlebrush plant (Callistemon citrinus). It became clear that neither the shade nor the litterfall of these plants was responsible for the suppression of plant and weed growth. Rather, a substance – which was identified as leptospermone – in the soil under the bottlebrush plant was shown to have bleaching activity on the emerging plants. The allelochemical leptospermone was extracted from the bottlebrush plant and chemically characterized. Leptospermone belongs to the triketone family and inhibits chloroplast development due to a lack of plastoquinone secondary to hepatic 4-hydroxyphenylpyruvate dioxygenase (HPPD) inhibition; thus, it served as a blueprint for the synthesis of nitisinone.[9]
In 1932, Grace Medes first described "a new disorder of tyrosine metabolism," She coined the condition "tyrosinosis" after observing 4-hydroxyphenylpyruvate in the urine of a 49-year-old man with myasthenia gravis. She proposed that the metabolic defect in this patient was a deficiency of 4-hydroxyphenylpyruvate dioxygenase, but her case remains puzzling and has since been assigned a separate OMIM number. The first typical patient with hepatorenal tyrosinemia was described in 1956 by Margaret D Baber at Edgware General Hospital in Middlesex, England. Starting the following year, Kiyoshi Sakai and colleagues, at the Jikei University School of Medicine in Tokyo, published 3 reports describing the clinical, biochemical, and pathological findings of a 2-year-old boy with hepatorenal tyrosinemia who was then thought to have an "atypical" case of tyrosinosis. Between 1963 and 1965, Swedish pediatrician Rolf Zetterström and colleagues at the Karolinska Institute in Sweden published the first detailed clinical account of hepatorenal tyrosinemia and its variants. Shortly thereafter, a Canadian group also described the clinical and laboratory findings of hepatorenal tyrosinemia. Both the Scandinavian and Canadian groups suggested that the Japanese patients described earlier by Sakai and colleagues had the same disorder, ie, hepatorenal tyrosinemia. In 1965, doubts emerged that the underlying biochemical cause of hepatorenal tyrosinemia was a defective form of the 4-hydroxyphenylpyruvate dioxygenase enzyme. In 1977, Bengt Lindblad and colleagues at the University of Gothenburg in Sweden demonstrated that the actual defect in causing hepatorenal tyrosinemia involved the fumarylacetoacetate hydrolase enzyme. This was subsequently confirmed using direct enzyme assays.[14]
Research directions
As of April 2020, two new clinical trials,[15] are underway in the USA for a Mass Spectrometry-based biomarker for the early and sensitive diagnosis of Tyrosinemia type 1 from blood plasma.
References
- de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, et al. (January 2013). "Recommendations for the management of tyrosinaemia type 1". Orphanet Journal of Rare Diseases. 8: 8. doi:10.1186/1750-1172-8-8. PMC 3558375. PMID 23311542.
- Chakrapani A, Holme E (2006). "Disorders of Tyrosine Metabolism". In Fernandes J, Saudubray J, van den Berghe G, Walter JH (eds.). Inborn Metabolic Diseases: Diagnosis and Treatment. Springer. pp. 233–243. doi:10.1007/978-3-540-28785-8_18. ISBN 978-3-540-28785-8.
- Paradis K (October 1996). "Tyrosinemia: the Quebec experience". Clinical and Investigative Medicine. 19 (5): 311–6. PMID 8889268.
- Enns GM, Packman S (2001). "Diagnosing Inborn Errors of Metabolism in the Newborn: Clinical Features" (PDF). NeoReviews. 2 (8): e183–e191. doi:10.1542/neo.2-8-e183. ISSN 1526-9906.
- "Clinical Review Report: Nitisinone (Orfadin): (Sobi Canada Inc.): Indication: For the treatment of patients with hereditary tyrosinemia type 1 in combination with dietary restriction of tyrosine and phenylalanine". CADTH Common Drug Reviews. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. 2018. PMID 30457777.
- "Physician's Guide to Tyrosinemia Type 1" (PDF). National Organization for Rare Disorders. Archived from the original (PDF) on 2014-02-11.
- "Tyrosine - structure, properties, function, benefits". aminoacidsguide.com. Retrieved 2020-05-02.
- Thimm E, Richter-Werkle R, Kamp G, Molke B, Herebian D, Klee D, et al. (March 2012). "Neurocognitive outcome in patients with hypertyrosinemia type I after long-term treatment with NTBC". Journal of Inherited Metabolic Disease. 35 (2): 263–8. doi:10.1007/s10545-011-9394-5. PMID 22069142. S2CID 23783926.
- Das AM (2017-07-24). "Clinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1)". The Application of Clinical Genetics. 10: 43–48. doi:10.2147/TACG.S113310. PMC 5533484. PMID 28769581.
- Lock EA, Ellis MK, Gaskin P, Robinson M, Auton TR, Provan WM, et al. (August 1998). "From toxicological problem to therapeutic use: the discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its toxicology and development as a drug". Journal of Inherited Metabolic Disease. 21 (5): 498–506. doi:10.1023/a:1005458703363. PMID 9728330. S2CID 6717818.
- "Tyrosinemia Type 1". NORD (National Organization for Rare Disorders). Retrieved 2020-05-01.
- Grompe M, St-Louis M, Demers SI, al-Dhalimy M, Leclerc B, Tanguay RM (August 1994). "A single mutation of the fumarylacetoacetate hydrolase gene in French Canadians with hereditary tyrosinemia type I". The New England Journal of Medicine. 331 (6): 353–7. doi:10.1056/NEJM199408113310603. PMID 8028615.
- Laberge C, Dallaire L (October 1967). "Genetic aspects of tyrosinemia in the Chicoutimi region". Canadian Medical Association Journal. 97 (18): 1099–101. PMC 1923580. PMID 6057677.
- "Hepatorenal Tyrosinemia". MedLink Neurology. Retrieved 2020-05-01.
- "Tyrosinemia Clinical Trials". wcg CenterWatch.
External links
- Reference, Genetics Home. "Tyrosinemia". Genetics Home Reference. Retrieved 2020-05-01.
- "Tyrosinemia Type 1". NORD (National Organization for Rare Disorders). Retrieved 2020-05-01.
| Classification | |
|---|---|
| External resources |