Mannich reaction
The Mannich reaction is an organic reaction which consists of an amino alkylation of an acidic proton placed next to a carbonyl functional group by formaldehyde and a primary or secondary amine or ammonia. The final product is a β-amino-carbonyl compound also known as a Mannich base.[1] Reactions between aldimines and α-methylene carbonyls are also considered Mannich reactions because these imines form between amines and aldehydes. The reaction is named after chemist Carl Mannich.[2][3]

| Mannich reaction | |
|---|---|
| Named after | Carl Mannich |
| Reaction type | Coupling reaction |
| Identifiers | |
| Organic Chemistry Portal | mannich-reaction |
| RSC ontology ID | RXNO:0000032 |
The Mannich reaction is an example of nucleophilic addition of an amine to a carbonyl group followed by dehydration to the Schiff base. The Schiff base is an electrophile which reacts in the second step in an electrophilic addition with a compound containing an acidic proton (which is, or had become an enol). The Mannich reaction is also considered a condensation reaction.
In the Mannich reaction, primary or secondary amines or ammonia, are employed for the activation of formaldehyde. Tertiary amines lack an N–H proton to form the intermediate enamine. α-CH-acidic compounds (nucleophiles) include carbonyl compounds, nitriles, acetylenes, aliphatic nitro compounds, α-alkyl-pyridines or imines. It is also possible to use activated phenyl groups and electron-rich heterocycles such as furan, pyrrole, and thiophene. Indole is a particularly active substrate; the reaction provides gramine derivatives.
When rationalizing the Mannich reaction, it can be clearly understood to be a mixed-Aldol reaction, dehydration of the alcohol, and conjugate addition of an amine (Michael reaction) all happening in "one-pot". Double Mannich reactions are also very common to set-up.
Reaction mechanism
The mechanism of the Mannich reaction starts with the formation of an iminium ion from the amine and the formaldehyde. Please note, the mechanism shown below is NOT correct. The pKa of the protonated oxygen is approximately -2. The amine base would simply deprotonate the carbonyl and stop the reaction. Consequently, it is imperative that this reaction is performed at a pH of approximately 4-5. The correct mechanism should start with a nucleophilic attack by the nitrogen atom on the carbonyl carbon.

The compound with the carbonyl functional group (in this case a ketone) can tautomerize to the enol form, after which it can attack the iminium ion.
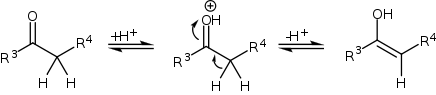

On methyl ketones, the enolization and the Mannich addition can occur twice, followed by an β-elimination to yield β-amino enone derivatives.[4][5]
Asymmetric Mannich reactions
Progress has been made towards asymmetric Mannich reactions. When properly functionalized the newly formed ethylene bridge in the Mannich adduct has two prochiral centers giving rise to two diastereomeric pairs of enantiomers. The first asymmetric Mannich reaction with an unmodified aldehyde was carried with (S)-proline as a naturally occurring chiral catalyst.[6]

The reaction taking place is between a simple aldehyde, such as propionaldehyde, and an imine derived from ethyl glyoxylate and p-methoxyaniline (PMP = paramethoxyphenyl) catalyzed by (S)-proline in dioxane at room temperature. The reaction product is diastereoselective with a preference for the syn-Mannich reaction 3:1 when the alkyl substituent on the aldehyde is a methyl group or 19:1 when the alkyl group the much larger pentyl group. Of the two possible syn adducts (S,S) or (R,R) the reaction is also enantioselective with a preference for the (S,S) adduct with enantiomeric excess larger than 99%. This stereoselectivity is explained in the scheme below.

Proline enters a catalytic cycle by reacting with the aldehyde to form an enamine. The two reactants (imine and enamine) line up for the Mannich reaction with Si facial attack of the imine by the Si-face of the enamine-aldehyde. Relief of steric strain dictates that the alkyl residue R of the enamine and the imine group are antiperiplanar on approach which locks in the syn mode of addition. The enantioselectivity is further controlled by hydrogen bonding between the proline carboxyl group and the imine. The transition state for the addition is a nine-membered ring with chair conformation with partial single bonds and double bonds. The proline group is converted back to the aldehyde and a single (S,S) isomer is formed.
By modification of the proline catalyst to it is also possible to obtain anti-Mannich adducts.[7]

An additional methyl group attached to proline forces a specific enamine approach and the transition state now is a 10-membered ring with addition in anti-mode. The diastereoselectivity is at least anti:syn 95:5 regardless of alkyl group size and the (S,R) enantiomer is preferred with at least 97% enantiomeric excess.
Applications
The Mannich reaction is used in many areas of organic chemistry, Examples include:
- alkyl amines
- peptides, nucleotides, antibiotics, and alkaloids (e.g. tropinone)
- agrochemicals, such as plant growth regulators[8]
- polymers
- catalysts
- Formaldehyde tissue crosslinking
- Pharmaceutical drugs (e.g. rolitetracycline (the Mannich product of tetracycline and pyrrolidine), fluoxetine (antidepressant), tramadol and tolmetin (anti-inflammatory drug).
- soap and detergents. These compounds are used in a variety of cleaning applications, automotive fuel treatments, and epoxy coatings
- polyetheramines from substituted branched chain alkyl ethers[9]
- α,β-unsaturated ketones by the thermal degradation of Mannich reaction products (e.g. methyl vinyl ketone from 1-diethylamino-butan-3-one)[10][11]
References
- Original translated from German Wiki
- Carl Mannich; Krösche, W. (1912). "Ueber ein Kondensationsprodukt aus Formaldehyd, Ammoniak und Antipyrin". Archiv der Pharmazie (in German). 250 (1): 647–667. doi:10.1002/ardp.19122500151. S2CID 94217627.
- Blicke, F. F. (2011). "The Mannich Reaction". Organic Reactions. 1 (10): 303–341. doi:10.1002/0471264180.or001.10. ISBN 978-0471264187.
- Cromwell, Norman H.; Soriano, David S.; Doomes, Earl (November 1980). "Mobile keto allyl systems. 18. Synthesis and chemistry of N-substituted and N,N-disubstituted 2-benzoyl-1-amino-3-propenes". The Journal of Organic Chemistry. 45 (24): 4983–4985. doi:10.1021/jo01312a034.
- Girreser, Ulrich; Heber, Dieter; Schütt, Martin (May 1998). "A Facile One-Pot Synthesis of 1-Aryl-2-(dimethylaminomethyl)prop-2-en-1-ones from Aryl Methyl Ketones". Synthesis. 1998 (5): 715–717. doi:10.1055/s-1998-2056.
- Córdova, A.; Watanabe, S.-I.; Tanaka, F.; Notz, W.; Barbas, C. F. (2002). "A highly enantioselective route to either enantiomer of both α- and β-amino acid derivatives". Journal of the American Chemical Society. 124 (9): 1866–1867. doi:10.1021/ja017833p. PMID 11866595.
- Mitsumori, S.; Zhang, H.; Cheong, P. H.-Y.; Houk, K.; Tanaka, F.; Barbas, C. F. (2006). "Direct asymmetric anti-Mannich-type reactions catalyzed by a designed amino acid". Journal of the American Chemical Society. 128 (4): 1040–1041. doi:10.1021/ja056984f. PMC 2532695. PMID 16433496.
- da Rosa, F. A. F.; Rebelo, R. A.; Nascimento, M. G. (2003). "Synthesis of new indolecarboxylic acids related to the plant hormone indoleacetic acid" (PDF). Journal of the Brazilian Chemical Society. 14 (1): 11–15. doi:10.1590/S0103-50532003000100003.
- Siegel, H.; Eggersdorfer, M. "Ketones". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a15_077.
- Wilds, A. L.; Nowak, R. M.; McCaleb, K. E. (1957). "1-Diethylamino-3-butanone (2-Butanone, 4-diethylamino-)". Organic Syntheses. 37: 18. doi:10.15227/orgsyn.037.0018.; Collective Volume, 4, p. 281
External links
| Authority control |
|---|