Nicotinic acetylcholine receptor
Nicotinic acetylcholine receptors, or nAChRs, are receptor polypeptides that respond to the neurotransmitter acetylcholine. Nicotinic receptors also respond to drugs such as the agonist nicotine. They are found in the central and peripheral nervous system, muscle, and many other tissues of many organisms. At the neuromuscular junction they are the primary receptor in muscle for motor nerve-muscle communication that controls muscle contraction. In the peripheral nervous system: (1) they transmit outgoing signals from the presynaptic to the postsynaptic cells within the sympathetic and parasympathetic nervous system, and (2) they are the receptors found on skeletal muscle that receive acetylcholine released to signal for muscular contraction. In the immune system, nAChRs regulate inflammatory processes and signal through distinct intracellular pathways.[1] In insects, the cholinergic system is limited to the central nervous system.[2]
The nicotinic receptors are considered cholinergic receptors, since they respond to acetylcholine. Nicotinic receptors get their name from nicotine which does not stimulate the muscarinic acetylcholine receptors but selectively binds to the nicotinic receptors instead.[3][4][5] The muscarinic acetylcholine receptor likewise gets its name from a chemical that selectively attaches to that receptor — muscarine.[6] Acetylcholine itself binds to both muscarinic and nicotinic acetylcholine receptors.[7]
As ionotropic receptors, nAChRs are directly linked to ion channels. New evidence suggests that these receptors can also use second messengers (as metabotropic receptors do) in some cases.[8] Nicotinic acetylcholine receptors are the best-studied of the ionotropic receptors.[3]
Since nicotinic receptors help transmit outgoing signals for the sympathetic and parasympathetic systems, nicotinic receptor antagonists such as hexamethonium interfere with the transmission of these signals. Thus, for example, nicotinic receptor antagonists interfere with the baroreflex that normally corrects changes in blood pressure by sympathetic and parasympathetic stimulation of the heart.
Structure
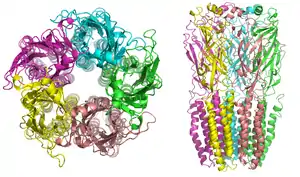
Nicotinic receptors, with a molecular mass of 290 kDa,[9] are made up of five subunits, arranged symmetrically around a central pore.[3] Each subunit comprises four transmembrane domains with both the N- and C-terminus located extracellularly. They possess similarities with GABAA receptors, glycine receptors, and the type 3 serotonin receptors (which are all ionotropic receptors), or the signature Cys-loop proteins.[10]
In vertebrates, nicotinic receptors are broadly classified into two subtypes based on their primary sites of expression: muscle-type nicotinic receptors and neuronal-type nicotinic receptors. In the muscle-type receptors, found at the neuromuscular junction, receptors are either the embryonic form, composed of α1, β1, γ, and δ subunits in a 2:1:1:1 ratio, or the adult form composed of α1, β1, δ, and ε subunits in a 2:1:1:1 ratio.[3][4][5][11] The neuronal subtypes are various homomeric (all one type of subunit) or heteromeric (at least one α and one β) combinations of twelve different nicotinic receptor subunits: α2−α10 and β2−β4. Examples of the neuronal subtypes include: (α4)3(β2)2, (α4)2(β2)3, (α3)2(β4)3, α4α6β3(β2)2, (α7)5, and many others. In both muscle-type and neuronal-type receptors, the subunits are very similar to one another, especially in the hydrophobic regions.
A number of electron microscopy and x-ray crystallography studies have provided very high resolution structural information for muscle and neuronal nAChRs and their binding domains.[9][12][13][14]
Binding to the receptor
As with all ligand-gated ion channels, opening of the nAChR channel pore requires the binding of a chemical messenger. Several different terms are used to refer to the molecules that bind receptors, such as ligand, agonist, or transmitter. As well as the endogenous agonist acetylcholine, agonists of the nAChR include nicotine, epibatidine, and choline. Nicotinic antagonists that block the receptor include mecamylamine, dihydro-β-erythroidine, and hexamethonium.
In muscle-type nAChRs, the acetylcholine binding sites are located at the α and either ε or δ subunits interface. In neuronal nAChRs, the binding site is located at the interface of an α and a β subunit or between two α subunits in the case of α7 receptors. The binding site is located in the extracellular domain near the N terminus.[4][15] When an agonist binds to the site, all present subunits undergo a conformational change and the channel is opened[16] and a pore with a diameter of about 0.65 nm opens.[4]
Opening the channel
Nicotinic AChRs may exist in different interconvertible conformational states. Binding of an agonist stabilises the open and desensitised states. In normal physiological conditions, the receptor needs exactly two molecules of ACh to open.[17] Opening of the channel allows positively charged ions to move across it; in particular, sodium enters the cell and potassium exits. The net flow of positively charged ions is inward.
The nAChR is a non-selective cation channel, meaning that several different positively charged ions can cross through.[3] It is permeable to Na+ and K+, with some subunit combinations that are also permeable to Ca2+.[4][18][19] The amount of sodium and potassium the channels allow through their pores (their conductance) varies from 50–110 pS, with the conductance depending on the specific subunit composition as well as the permeant ion.[20]
Many neuronal nAChRs can affect the release of other neurotransmitters.[5] The channel usually opens rapidly and tends to remain open until the agonist diffuses away, which usually takes about 1 millisecond.[4] However, AChRs can spontaneously open with no ligands bound or can spontaneously close with ligands bound, and mutations in the channel can shift the likelihood of either event.[21][16] Therefore, ACh binding changes the probability of pore opening, which increases as more ACh binds.
The nAChR is unable to bind ACh when bound to any of the snake venom α-neurotoxins. These α-neurotoxins antagonistically bind tightly and noncovalently to nAChRs of skeletal muscles and in neurons, thereby blocking the action of ACh at the postsynaptic membrane, inhibiting ion flow and leading to paralysis and death. The nAChR contains two binding sites for snake venom neurotoxins. Progress towards discovering the dynamics of binding action of these sites has proved difficult, although recent studies using normal mode dynamics[22] have aided in predicting the nature of both the binding mechanisms of snake toxins and of ACh to nAChRs. These studies have shown that a twist-like motion caused by ACh binding is likely responsible for pore opening, and that one or two molecules of α-bungarotoxin (or other long-chain α-neurotoxin) suffice to halt this motion. The toxins seem to lock together neighboring receptor subunits, inhibiting the twist and therefore, the opening motion.[23]
Effects
The activation of receptors by nicotine modifies the state of neurons through two main mechanisms. On one hand, the movement of cations causes a depolarization of the plasma membrane (which results in an excitatory postsynaptic potential in neurons) leading to the activation of voltage-gated ion channels. On the other hand, the entry of calcium acts, either directly or indirectly, on different intracellular cascades. This leads, for example, to the regulation of the activity of some genes or the release of neurotransmitters.
Receptor regulation
Receptor desensitisation
Ligand-bound desensitisation of receptors was first characterised by Katz and Thesleff in the nicotinic acetylcholine receptor.[24]
Prolonged or repeated exposure to a stimulus often results in decreased responsiveness of that receptor toward a stimulus, termed desensitisation. nAChR function can be modulated by phosphorylation[25] by the activation of second messenger-dependent protein kinases. PKA[24] and PKC,[26] as well as tyrosine kinases,[27] have been shown to phosphorylate the nAChR resulting in its desensitisation. It has been reported that, after prolonged receptor exposure to the agonist, the agonist itself causes an agonist-induced conformational change in the receptor, resulting in receptor desensitisation.[28]
Desensitised receptors can revert to a prolonged open state when an agonist is bound in the presence of a positive allosteric modulator, for example PNU-120596.[29] Also, there is evidence that indicates specific chaperone molecules have regulatory effects on these receptors.[30]
Roles
The subunits of the nicotinic receptors belong to a multigene family (16 members in humans) and the assembly of combinations of subunits results in a large number of different receptors (for more information see the Ligand-Gated Ion Channel database). These receptors, with highly variable kinetic, electrophysiological and pharmacological properties, respond to nicotine differently, at very different effective concentrations. This functional diversity allows them to take part in two major types of neurotransmission. Classical synaptic transmission (wiring transmission) involves the release of high concentrations of neurotransmitter, acting on immediately neighboring receptors. In contrast, paracrine transmission (volume transmission) involves neurotransmitters released by synaptic boutons, which then diffuse through the extra-cellular medium until they reach their receptors, which may be distant.[31] Nicotinic receptors can also be found in different synaptic locations; for example the muscle nicotinic receptor always functions post-synaptically. The neuronal forms of the receptor can be found both post-synaptically (involved in classical neurotransmission) and pre-synaptically[32] where they can influence the release of multiple neurotransmitters.
Subunits
17 vertebrate nAChR subunits have been identified, which are divided into muscle-type and neuronal-type subunits. However, although an α8 subunit/gene is present in avian species such as the chicken, it is not present in human or mammalian species.[33]
The nAChR subunits have been divided into 4 subfamilies (I-IV) based on similarities in protein sequence.[34] In addition, subfamily III has been further divided into 3 types.
| Neuronal-type | Muscle-type | ||||
| I | II | III | IV | ||
|---|---|---|---|---|---|
| α9, α10 | α7, α8 | 1 | 2 | 3 | α1, β1, δ, γ, ε |
| α2, α3, α4, α6 | β2, β4 | β3, α5 | |||
- α genes: CHRNA1 (muscle), CHRNA2 (neuronal), CHRNA3, CHRNA4, CHRNA5, CHRNA6, CHRNA7, CHRNA8, CHRNA9, CHRNA10
- β genes: CHRNB1 (muscle), CHRNB2 (neuronal), CHRNB3, CHRNB4
- Other genes: CHRND (delta), CHRNE (epsilon), CHRNG (gamma)
Neuronal nAChRs are transmembrane proteins that form pentameric structures assembled from a family of subunits composed of α2-α10 and β2-β4.[35] These subunits were discovered from the mid-1980s through the early 1990s, when cDNAs for multiple nAChR subunits were cloned from rat and chicken brains, leading to the identification of eleven different genes (twelve in chickens) that code for neuronal nAChR subunits; The subunit genes identified were named α2–α10 (α8 only found in chickens) and β2–β4.[36] It has also been discovered that various subunit combinations could form functional nAChRs that could be activated by acetylcholine and nicotine, and the different combinations of subunits generate subtypes of nAChRs with diverse functional and pharmacological properties.[37] When expressed alone, α7, α8, α9, and α10 are able to form functional receptors, but other α subunits require the presence of β subunits to form functional receptors.[35] In mammals, nAchR subunits have been found to be encoded by 17 genes, and of these, nine genes encoding α-subunits and three encoding β-subunits are expressed in the brain. β2 subunit-containing nAChRs (β2nAChRs) and α7nAChRs are widely expressed in the brain, whereas other nAChR subunits have more restricted expression.[38]
CHRNA5/A3/B4
An important nAchR gene cluster (CHRNA5/A3/B4) contains the genes encoding for the α5, α3 and β4 subunits. Genetic studies have identified single nucleotide polymorphisms (SNPs) in the chromosomal locus encoding these three nAChR genes as risk factors for nicotine dependence, lung cancer, chronic obstructive pulmonary disease, alcoholism, and peripheral arterial disease.[35][39] The CHRNA5/A3/B4 nAChR subunit genes are found in a tight cluster in chromosomal region 15q24–25. The nAChR subunits encoded by this locus form the predominant nicotinic receptor subtypes expressed in the peripheral nervous system (PNS) and other key central nervous system (CNS) sites, such as the medial habenula, a structure between the limbic forebrain and midbrain involved in major cholinergic circuitry pathways.[35] Further research of the CHRNA5/A3/B4 genes have revealed that “neuronal” nAChR genes are also expressed in non-neuronal cells where they are involved in various fundamental processes, such as inflammation.[40] The CHRNA5/A3/B4 genes are co-expressed in many cell types and the transcriptional activities of the promoter regions of the three genes are regulated by many of the same transcription factors, demonstrating that their clustering may reflect control of gene expression.[35]
CHRNA6/CHRNB3
CHRNB3 and CHRNA6 are also grouped in a gene cluster, located on 8p11.[39] Multiple studies have shown that SNPS in the CHRNB3–CHRNA6 have been linked to nicotine dependence and smoking behavior, such as two SNPs in CHRNB3, rs6474413 and rs10958726.[39] Genetic variation in this region also displays influence susceptibility to use drugs of abuse, including cocaine and alcohol consumption.[41] Nicotinic receptors containing α6 or β3 subunits expressed in brain regions, especially in the ventral tegmental area and substantia nigra, are important for drug behaviors due to their role in dopamine release.[42] Genetic variation in these genes can alter sensitivity to drugs of abuse in numerous ways, including changing the amino acid structure of the protein or cause alterations in transcriptional and translational regulation.[41]
CHRNA4/CHRNB2
Other well studied nAChR genes include the CHRNA4 and CHRNB2, which have been associated as Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE) genes.[39][43] Both of these nAChR subunits are present in the brain and the occurrence of mutations in these two subunits cause a generalized type of epilepsy. Examples include the CHRNA4 insertion mutation 776ins3 that is associated with nocturnal seizures and psychiatric disorders, and the CHRNB2 mutation I312M that seems to cause not only epilepsy but also very specific cognitive deficits, such as deficits in learning and memory.[43][44] There is naturally occurring genetic variation between these two genes and analysis of single nucleotide polymorphisms (SNPs) and other gene modifications show a higher variation in the CHRNA4 gene than in the CHRNB2 gene, implying that nAChR β2, the protein encoded by CHRNB2, associates with more subunits than α4. CHRNA2 has also been reported as a third candidate for nocturnal frontal lobe seizures.[39][43]
CHRNA7
Several studies have reported an association between CHRNA7 and endophenotypes of psychiatric disorders and nicotine dependence, contributing to the significant clinical relevance of α7 and research being done on it.[43] CHRNA7 was one of the first genes that had been considered to be involved with schizophrenia. Studies identified several CHRNA7 promoter polymorphisms that reduce the genes transcriptional activity to be associated with schizophrenia, which is consistent with the finding of reduced levels of a7 nAChRs in the brain of schizophrenic patients.[43] Both nAChRs subtypes, α4β2 and α7, have been found to be significantly reduced in post-mortem studies of individuals with schizophrenia.[45] Additionally, smoking rates are significantly higher in those with schizophrenia, implying that smoking nicotine may be a form of self-medicating.[46]
Notable variations
Nicotinic receptors are pentamers of these subunits; i.e., each receptor contains five subunits. Thus, there is immense potential of variation of these subunits. However, some of them are more commonly found than others. The most broadly expressed subtypes include (α1)2β1δε (adult muscle-type), (α3)2(β4)3 (ganglion-type), (α4)2(β2)3 (CNS-type) and (α7)5 (another CNS-type).[47] A comparison follows:
| Receptor-type | Location | Effect; functions | Nicotinic agonists | Nicotinic antagonists |
|---|---|---|---|---|
| Muscle-type: (α1)2β1δε[47] or (α1)2β1δγ |
Neuromuscular junction | EPSP, mainly by increased Na+ and K+ permeability | ||
| Ganglion-type: (α3)2(β4)3 |
autonomic ganglia | EPSP, mainly by increased Na+ and K+ permeability | ||
| Heteromeric CNS-type: (α4)2(β2)3 |
Brain | Post- and presynaptic excitation,[47] mainly by increased Na+ and K+ permeability. Major subtype involved in the attention-enhancing and rewarding effects of nicotine as well as the pathophysiology of nicotine addiction.[49][50][51] |
| |
| Further CNS-type: (α3)2(β4)3 |
Brain | Post- and presynaptic excitation | ||
| Homomeric CNS-type: (α7)5 |
Brain | Post- and presynaptic excitation,[47] mainly by increased Na+, K+ and Ca2+ permeability. Major subtype involved in some of the cognitive effects of nicotine.[52] Moreover, activation of (α7)5 could improve neurovascular coupling response in neurodegenerative disease[53] and neurogenesis in ischemic stroke.[54] Also involved in the pro-angiogenic effects of nicotine and accelerate the progression of chronic kidney disease in smokers.[55][56][57] | ||
See also
References
- Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, et al. (August 2014). "α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release". Molecular Medicine. 20 (1): 350–8. doi:10.2119/molmed.2013.00117. PMC 4153835. PMID 24849809.
- Yamamoto I (1999). "Nicotine to Nicotinoids: 1962 to 1997". Nicotinoid Insecticides and the Nicotinic Acetylcholine Receptor. pp. 3–27. doi:10.1007/978-4-431-67933-2_1. ISBN 978-4-431-68011-6.
- Purves D, Augustine GJ, Fitzpatrick D, Hall WC, LaMantia A, McNamara JO, White LE (2008). Neuroscience (4th ed.). Sinauer Associates. pp. 122–6. ISBN 978-0-87893-697-7.
- Siegel GJ, Agranoff BW, Fisher SK, Albers RW, Uhler MD (1999). "Basic Neurochemistry: Molecular, Cellular and Medical Aspects". GABA Receptor Physiology and Pharmacology (6th ed.). American Society for Neurochemistry. Retrieved 2008-10-01.
- Itier V, Bertrand D (August 2001). "Neuronal nicotinic receptors: from protein structure to function". FEBS Letters. 504 (3): 118–25. doi:10.1016/s0014-5793(01)02702-8. PMID 11532443.
- Ishii M, Kurachi Y (1 October 2006). "Muscarinic acetylcholine receptors". Current Pharmaceutical Design. 12 (28): 3573–81. doi:10.2174/138161206778522056. PMID 17073660.
- Lott EL, Jones EB (2020). "Cholinergic Toxicity". StatPearls. StatPearls Publishing. PMID 30969605.
- Kabbani N, Nordman JC, Corgiat BA, Veltri DP, Shehu A, Seymour VA, Adams DJ (December 2013). "Are nicotinic acetylcholine receptors coupled to G proteins?". BioEssays. 35 (12): 1025–34. doi:10.1002/bies.201300082. PMID 24185813.
- Unwin N (March 2005). "Refined structure of the nicotinic acetylcholine receptor at 4A resolution". Journal of Molecular Biology. 346 (4): 967–89. doi:10.1016/j.jmb.2004.12.031. PMID 15701510.
- Cascio M (May 2004). "Structure and function of the glycine receptor and related nicotinicoid receptors". The Journal of Biological Chemistry. 279 (19): 19383–6. doi:10.1074/jbc.R300035200. PMID 15023997.
- Giniatullin R, Nistri A, Yakel JL (July 2005). "Desensitization of nicotinic ACh receptors: shaping cholinergic signaling". Trends in Neurosciences. 28 (7): 371–8. doi:10.1016/j.tins.2005.04.009. PMID 15979501.
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK (May 2001). "Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors". Nature. 411 (6835): 269–76. Bibcode:2001Natur.411..269B. doi:10.1038/35077011. PMID 11357122.
- Zouridakis M, Giastas P, Zarkadas E, Chroni-Tzartou D, Bregestovski P, Tzartos SJ (November 2014). "Crystal structures of free and antagonist-bound states of human α9 nicotinic receptor extracellular domain". Nature Structural & Molecular Biology. 21 (11): 976–80. doi:10.1038/nsmb.2900. PMID 25282151.
- Morales-Perez CL, Noviello CM, Hibbs RE (October 2016). "X-ray structure of the human α4β2 nicotinic receptor". Nature. 538 (7625): 411–415. Bibcode:2016Natur.538..411M. doi:10.1038/nature19785. PMC 5161573. PMID 27698419.
- Squire L (2003). Fundamental neuroscience (2nd ed.). Amsterdam: Acad. Press. p. 1426. ISBN 978-0-12-660303-3.
- Colquhoun D, Sivilotti LG (June 2004). "Function and structure in glycine receptors and some of their relatives". Trends in Neurosciences. 27 (6): 337–44. CiteSeerX 10.1.1.385.3809. doi:10.1016/j.tins.2004.04.010. PMID 15165738.
- Aidley DJ (1998). The physiology of excitable cells (4th ed.). Cambridge, UK: Cambridge University Press. ISBN 978-0521574150. OCLC 38067558.
- Beker F, Weber M, Fink RH, Adams DJ (September 2003). "Muscarinic and nicotinic ACh receptor activation differentially mobilize Ca2+ in rat intracardiac ganglion neurons". Journal of Neurophysiology. 90 (3): 1956–64. doi:10.1152/jn.01079.2002. PMID 12761283.
- Weber M, Motin L, Gaul S, Beker F, Fink RH, Adams DJ (January 2005). "Intravenous anaesthetics inhibit nicotinic acetylcholine receptor-mediated currents and Ca2+ transients in rat intracardiac ganglion neurons". British Journal of Pharmacology. 144 (1): 98–107. doi:10.1038/sj.bjp.0705942. PMC 1575970. PMID 15644873.
- Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, et al. (May 1986). "Molecular distinction between fetal and adult forms of muscle acetylcholine receptor". Nature. 321 (6068): 406–11. Bibcode:1986Natur.321..406M. doi:10.1038/321406a0. PMID 2423878.
- Einav T, Phillips R (April 2017). "Monod-Wyman-Changeux Analysis of Ligand-Gated Ion Channel Mutants". The Journal of Physical Chemistry B. 121 (15): 3813–3824. arXiv:1701.06122. Bibcode:2017arXiv170106122E. doi:10.1021/acs.jpcb.6b12672. PMC 5551692. PMID 28134524.
- Levitt M, Sander C, Stern PS (February 1985). "Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme". Journal of Molecular Biology. 181 (3): 423–47. doi:10.1016/0022-2836(85)90230-x. PMID 2580101.
- Samson AO, Levitt M (April 2008). "Inhibition mechanism of the acetylcholine receptor by alpha-neurotoxins as revealed by normal-mode dynamics". Biochemistry. 47 (13): 4065–70. doi:10.1021/bi702272j. PMC 2750825. PMID 18327915.
- Pitchford S, Day JW, Gordon A, Mochly-Rosen D (November 1992). "Nicotinic acetylcholine receptor desensitization is regulated by activation-induced extracellular adenosine accumulation". The Journal of Neuroscience. 12 (11): 4540–4. doi:10.1523/JNEUROSCI.12-11-04540.1992. PMC 6576003. PMID 1331363.
- Huganir RL, Greengard P (February 1983). "cAMP-dependent protein kinase phosphorylates the nicotinic acetylcholine receptor". Proceedings of the National Academy of Sciences of the United States of America. 80 (4): 1130–4. Bibcode:1983PNAS...80.1130H. doi:10.1073/pnas.80.4.1130. PMC 393542. PMID 6302672.
- Safran A, Sagi-Eisenberg R, Neumann D, Fuchs S (August 1987). "Phosphorylation of the acetylcholine receptor by protein kinase C and identification of the phosphorylation site within the receptor delta subunit". The Journal of Biological Chemistry. 262 (22): 10506–10. PMID 3038884.
- Hopfield JF, Tank DW, Greengard P, Huganir RL (December 1988). "Functional modulation of the nicotinic acetylcholine receptor by tyrosine phosphorylation". Nature. 336 (6200): 677–80. Bibcode:1988Natur.336..677H. doi:10.1038/336677a0. PMID 3200319.
- Barrantes FJ (September 1978). "Agonist-mediated changes of the acetylcholine receptor in its membrane environment". Journal of Molecular Biology. 124 (1): 1–26. doi:10.1016/0022-2836(78)90144-4. PMID 712829.
- Hurst RS, Hajós M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, et al. (April 2005). "A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization". The Journal of Neuroscience. 25 (17): 4396–405. doi:10.1523/JNEUROSCI.5269-04.2005. PMC 6725110. PMID 15858066.
- Sadigh-Eteghad S, Majdi A, Talebi M, Mahmoudi J, Babri S (May 2015). "Regulation of nicotinic acetylcholine receptors in Alzheimer׳s disease: a possible role of chaperones". European Journal of Pharmacology. 755: 34–41. doi:10.1016/j.ejphar.2015.02.047. PMID 25771456.
- Picciotto MR, Higley MJ, Mineur YS (October 2012). "Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior". Neuron. 76 (1): 116–29. doi:10.1016/j.neuron.2012.08.036. PMC 3466476. PMID 23040810.
- Wonnacott S (February 1997). "Presynaptic nicotinic ACh receptors". Trends in Neurosciences. 20 (2): 92–8. doi:10.1016/S0166-2236(96)10073-4. PMID 9023878.
- Graham A, Court JA, Martin-Ruiz CM, Jaros E, Perry R, Volsen SG, et al. (2002). "Immunohistochemical localisation of nicotinic acetylcholine receptor subunits in human cerebellum". Neuroscience. 113 (3): 493–507. doi:10.1016/S0306-4522(02)00223-3. PMID 12150770.
- Le Novère N, Changeux JP (February 1995). "Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells". Journal of Molecular Evolution. 40 (2): 155–72. Bibcode:1995JMolE..40..155L. doi:10.1007/BF00167110. PMID 7699721.
- Improgo MR, Scofield MD, Tapper AR, Gardner PD (October 2010). "The nicotinic acetylcholine receptor CHRNA5/A3/B4 gene cluster: dual role in nicotine addiction and lung cancer". Progress in Neurobiology. 92 (2): 212–26. doi:10.1016/j.pneurobio.2010.05.003. PMC 2939268. PMID 20685379.
- Tammimäki A, Horton WJ, Stitzel JA (October 2011). "Recent advances in gene manipulation and nicotinic acetylcholine receptor biology". Biochemical Pharmacology. 82 (8): 808–19. doi:10.1016/j.bcp.2011.06.014. PMC 3162071. PMID 21704022.
- Graham A, Court JA, Martin-Ruiz CM, Jaros E, Perry R, Volsen SG, et al. (September 2002). "Immunohistochemical localisation of nicotinic acetylcholine receptor subunits in human cerebellum". Neuroscience. 113 (3): 493–507. doi:10.1016/S0306-4522(02)00223-3. PMID 12150770.
- Changeux JP (June 2010). "Nicotine addiction and nicotinic receptors: lessons from genetically modified mice". Nature Reviews. Neuroscience. 11 (6): 389–401. doi:10.1038/nrn2849. PMID 20485364.
- Greenbaum L, Lerer B (October 2009). "Differential contribution of genetic variation in multiple brain nicotinic cholinergic receptors to nicotine dependence: recent progress and emerging open questions". Molecular Psychiatry. 14 (10): 912–45. doi:10.1038/mp.2009.59. PMID 19564872.
- Gahring LC, Rogers SW (January 2006). "Neuronal nicotinic acetylcholine receptor expression and function on nonneuronal cells". The AAPS Journal. 7 (4): E885-94. doi:10.1208/aapsj070486. PMC 2750958. PMID 16594641.
- Kamens HM, Corley RP, Richmond PA, Darlington TM, Dowell R, Hopfer CJ, et al. (September 2016). "Evidence for Association Between Low Frequency Variants in CHRNA6/CHRNB3 and Antisocial Drug Dependence". Behavior Genetics. 46 (5): 693–704. doi:10.1007/s10519-016-9792-4. PMC 4975622. PMID 27085880.
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ (October 2007). "The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum". Biochemical Pharmacology. 74 (8): 1235–46. doi:10.1016/j.bcp.2007.07.032. PMC 2735219. PMID 17825262.
- Steinlein OK, Bertrand D (November 2008). "Neuronal nicotinic acetylcholine receptors: from the genetic analysis to neurological diseases". Biochemical Pharmacology. 76 (10): 1175–83. doi:10.1016/j.bcp.2008.07.012. PMID 18691557.
- Bertrand D, Elmslie F, Hughes E, Trounce J, Sander T, Bertrand S, Steinlein OK (December 2005). "The CHRNB2 mutation I312M is associated with epilepsy and distinct memory deficits". Neurobiology of Disease. 20 (3): 799–804. doi:10.1016/j.nbd.2005.05.013. PMID 15964197.
- Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, et al. (October 2000). "Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia". Neuropsychopharmacology. 23 (4): 351–64. doi:10.1016/S0893-133X(00)00121-4. PMID 10989262.
- McLean SL, Grayson B, Idris NF, Lesage AS, Pemberton DJ, Mackie C, Neill JC (April 2011). "Activation of α7 nicotinic receptors improves phencyclidine-induced deficits in cognitive tasks in rats: implications for therapy of cognitive dysfunction in schizophrenia". European Neuropsychopharmacology. 21 (4): 333–43. doi:10.1016/j.euroneuro.2010.06.003. hdl:10454/8464. PMID 20630711.
- Rang HP (2003). Pharmacology (5th ed.). Edinburgh: Churchill Livingstone. ISBN 978-0-443-07145-4.
- Neurosci.pharm - MBC 3320 Acetylcholine Archived 2007-12-27 at the Wayback Machine
- Sarter M (August 2015). "Behavioral-Cognitive Targets for Cholinergic Enhancement". Current Opinion in Behavioral Sciences. 4: 22–26. doi:10.1016/j.cobeha.2015.01.004. PMC 5466806. PMID 28607947.
- Wu J, Gao M, Shen JX, Shi WX, Oster AM, Gutkin BS (October 2013). "Cortical control of VTA function and influence on nicotine reward". Biochemical Pharmacology. 86 (8): 1173–80. doi:10.1016/j.bcp.2013.07.013. PMID 23933294.
- "Nicotine: Biological activity". IUPHAR/BPS Guide to Pharmacology. International Union of Basic and Clinical Pharmacology. Retrieved 7 February 2016.
Kis as follows; α2β4=9900nM [5], α3β2=14nM [1], α3β4=187nM [1], α4β2=1nM [4,6]. Due to the heterogeneity of nACh channels we have not tagged a primary drug target for nicotine, although the α4β2 is reported to be the predominant high affinity subtype in the brain which mediates nicotine addiction [2-3].
- Levin ED (May 2012). "α7-Nicotinic receptors and cognition". Current Drug Targets. 13 (5): 602–6. doi:10.2174/138945012800398937. PMID 22300026.
- Sadigh-Eteghad S, Mahmoudi J, Babri S, Talebi M (November 2015). "Effect of alpha-7 nicotinic acetylcholine receptor activation on beta-amyloid induced recognition memory impairment. Possible role of neurovascular function". Acta Cirurgica Brasileira. 30 (11): 736–42. doi:10.1590/S0102-865020150110000003. PMID 26647792.
- Wang J, Lu Z, Fu X, Zhang D, Yu L, Li N, et al. (May 2017). "Alpha-7 Nicotinic Receptor Signaling Pathway Participates in the Neurogenesis Induced by ChAT-Positive Neurons in the Subventricular Zone". Translational Stroke Research. 8 (5): 484–493. doi:10.1007/s12975-017-0541-7. PMC 5704989. PMID 28551702.
- Lee J, Cooke JP (November 2012). "Nicotine and pathological angiogenesis". Life Sciences. 91 (21–22): 1058–64. doi:10.1016/j.lfs.2012.06.032. PMC 3695741. PMID 22796717.
- Jain G, Jaimes EA (October 2013). "Nicotine signaling and progression of chronic kidney disease in smokers". Biochemical Pharmacology. 86 (8): 1215–23. doi:10.1016/j.bcp.2013.07.014. PMC 3838879. PMID 23892062.
- Mihalak KB, Carroll FI, Luetje CW (September 2006). "Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors". Molecular Pharmacology. 70 (3): 801–5. doi:10.1124/mol.106.025130. PMID 16766716.
External links
| Wikiversity has learning resources about Poisson–Boltzmann profile for an ion channel |
 Media related to Nicotinic acetylcholine receptors at Wikimedia Commons
Media related to Nicotinic acetylcholine receptors at Wikimedia Commons- Calculated spatial position of Nicotinic acetylcholine receptor in the lipid bilayer