P-type calcium channel
The P-type calcium channel is a type of voltage-dependent calcium channel. Similar to many other high-voltage-gated calcium channels, the α1 subunit determines most of the channel's properties.[1] The 'P' signifies cerebellar Purkinje cells, referring to the channel's initial site of discovery.[2][3] P-type calcium channels play a similar role to the N-type calcium channel in neurotransmitter release at the presynaptic terminal and in neuronal integration in many neuronal types.
History
The calcium channel experiments that led to the discovery of P-type calcium channels were initially completed by Llinás and Sugimori in 1980.[2] P type calcium channels were named in 1989 because they were discovered within mammalian Purkinje neurons.[3] They were able to use an in vitro preparation to examine the ionic currents that account for Purkinje cells' electrophysiological properties. They found that there are calcium dependent action potentials which rise slowly and fall quickly then undergo hyperpolarization. The action potentials were voltage dependent and the afterhyperpolarizing potentials were connected to the spike bursts, located within the dendrites of the Purkinje cells. Without calcium flux in the Purkinje cells, action potentials fire sporadically at a high frequency.[2]
Basic features and structure
| calcium channel, voltage-dependent, P/Q type, alpha 1A subunit | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Symbol | CACNA1A | ||||||
| Alt. symbols | Cav2.1, CACNL1A4, SCA6, MHP1, MHP | ||||||
| IUPHAR | 532 | ||||||
| NCBI gene | 773 | ||||||
| HGNC | 1388 | ||||||
| OMIM | 601011 | ||||||
| RefSeq | NM_000068 | ||||||
| UniProt | O00555 | ||||||
| Other data | |||||||
| Locus | Chr. 19 p13 | ||||||
| |||||||
P-type calcium channels are voltage dependent calcium channels that are classified under the high voltage activated class channel, along with L-, N-, Q- and R-type channels. These channels require a strong depolarization in order to be activated. They are found at axon terminals, as well as in somatodendritic areas of neurons within the central and peripheral nervous system.[1] P-type calcium channels are also critical to vesicle release, specifically neurotransmitters and hormones[4] at synaptic terminals of excitatory and inhibitory synapses.[1]
Voltage gated P-type calcium channels consist of a main pore-forming α1 subunit (which is more specifically referred to as CaV2.1),[5] an α2 subunit and a β subunit. There can be γ subunits found in calcium channels of skeletal muscles.[6] The α1 subunit is encoded specifically by the CACNA1A gene[1] and is composed of four domains, each containing six transmembrane (S1-S6) spanning α helices. The S1-S2 loop and the S6 region are thought to be responsible for the channel's inactivation, the S4 region serves as the voltage sensor and S5-S6 loop forms the pore.[4] There are seven subunits within the α1 subunit. The A subunit, called α1ACa2+, corresponds to what is functionally defined as the P-type and Q-type isoforms. P-type and Q-type calcium channels are closely related as they are produced from the same gene via alternative splicing. As a complication of the alternative splicing, P-type and Q-type channels may have different subunit compositions.[6] The β subunit regulates the kinetics and expression of the channel, along with the α2δ subunit.[1]
Channel distribution
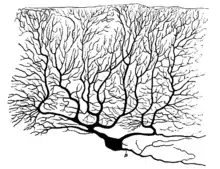
The majority of P-type calcium channels are located in the nervous system and heart. Antibody labeling is the primary method used to identify channel location.[7]
Areas of high expression in mammalian systems include:
- Purkinje cell dendrites[8]
- Smooth endoplasmic reticulum
- Plasma membrane of the soma (cell body)
- Periglomerular cells in the Olfactory bulb
- Cerebellar cortex
- Neurons in the brainstem, entorhinal and pyriform cortices, and the habenula.[7]
Channel blockers
P-type calcium channel blockers act to impede the flow of calcium. The blocking of calcium currents may cause the organism to experience impaired functioning and viability. These effects can lead to various diseases which are described in more detail in the section below.
The pore of P-type calcium channels are sensitive to compounds that can be divided into three groups:
- Peptide ion channel blockers
- Low molecular weight compounds
- Therapeutics [1]
There are only two peptide toxins that selectively block P-type channels: ω-agatoxin IVA and ω-agatoxin IVB. The other blockers mentioned, such as the low molecular weight and therapeutic blockers, are nonselective. This means they act can act on P-type channels as well as other channels.[1]
Selective peptide toxin ω-agatoxin

The two known blockers which are specific to P-type calcium channels are peptides derived from the spider venom of Agelenopsis aperta. The toxins from this venom which show selectivity for P-type channels are ω-agatoxin IVA and ω-agatoxin IVB. Each of these peptide toxins are made of 48 amino acids which are bound by four disulfide bonds. Although ω-agatoxin IVA and ω-agatoxin IVB have the same affinity and selectivity for P-type channels, their kinetics are different. The ω-agatoxin IVA effects the gating mechanism of the P-type channel. When there is a strong depolarization to activate the channel, ω-agatoxin IVA can no longer block the channel. Therefore, ω-agatoxin IVA has a very low affinity for the channel when it is open. It binds to the α1A subunit on the outside of the pore. The ω-agatoxin IVA receptor on the P-type channel is located at the S3-S4 linker. On the other hand, channel blocking by ω-agatoxin IVB occurs much more slowly. Yet, similar to ω-agatoxin IVA, ω-agatoxin IVB cannot bind to the channel upon a strong depolarization.[1]
Non-selective peptide toxins
- ω-Grammotoxin SIA is a peptide toxin derived from the venom of the spider Grammostola spatulata. It acts to modify the P-type channel gating.
- ω-PnTx3-3, PnTx3-3, and phonetoxin IIA are all toxins from the spider Phonoetrica nigriventer which act to block the current through the P-type calcium channels.
- DW13.3 is a peptide toxin from the spider Filistata hibernalis and it is composed of 74 amino acids. It also functions to block the current through P-type calcium channels.
- ω-Conotoxins are derived from the venom of cone snails. ω-Conotoxin MVIIC acts within the hippocampal CA1 pyramidal neurons to block the P-type channels. Also, within the hippocampal CA3 neurons, this toxin blocks synaptic transmission. Its effects are slow.
- Calcicludine is from venom of Dendroaspis angusticeps, which is a green mamba. It has the ability to voltage-dependently block P-type channels.
- Kurotoxin is from venom of the scorpion Parabuthus. In neurons in the thalamus, kurtoxin decreases high threshold calcium currents, however, in the Purkinje cells, it increases the calcium currents.[1]
Low molecular weight P-type channel blockers
Low molecular weight channel blockers have advantages over peptide blockers in drug development. One advantage of low molecular weight channel blockers is that they can penetrate tissue, which is important for crossing the blood-brain barrier. There is no specific low molecular weight channel blocker for P-type channels. However, there are a number of these blocker compounds which can effect the activity of the P-type channels.[1] These include:
- Roscovitine is an inhibitor of cyclin-dependent kinase. It increases the current of calcium in neostriatal interneurons by slowing the deactivation of the channel. Also, roscovitine can either act as an agonist or antagonist for the P-type calcium channels in the presynaptic membrane.
- Isoprenaline is a β-adrenoceptor agonist and it causes an increase in P-type calcium channel current. Isoprenaline acts through a cAMP signaling pathway.
- Eliprodil and antazoline are NMDA receptor antagonists and act to block P-type channels. Eliprodil can decrease P-type channel currents in the Purkinje neurons in the cerebellum.
- Dodecylamine can only block P-type channels when they are in the open state.
- Ethanol can block P-type channels when at a high enough concentration. The blocking of the P-type channels could be the reason for ataxia when drinking alcohol.[1]
Therapeutics
There are therapeutics used clinically which can effect the activity of P-type calcium channels. However, the primary target of these therapeutics are not thought to be P-type channels. For example, calcium antagonists, which are used to treat coronary heart disease, hypertension, and cardiac arrhythmia, act by inhibiting L-type or T-type calcium channels. Some of these calcium antagonists include verapamil, diltiazem, amlodipine, benidipine, cilnidipine, nicardipine, and barnidipine. Although their main target is not P-type channels, these calcium antagonists also act to block the function of P-type channels. Moreover, flunarizine is another calcium antagonist which is used to treat migraines. Its main targets are voltage-gated calcium channels and sodium channels. Flunarizine inhibits the P-type channels that are located in the neocortical slices. It works to inhibit the inward flux of calcium. The migraines that it helps to prevent are due to mutations within the "cacna1a" gene of the P-type channel subunit. Also, compounds that block P-type channels are shown to help with seizures. Epileptic seizures are caused by increased neurotransmission, which is partially a result of P-type channels. Compounds such as levetiracetam, lamotrigine, and carbamazepine are known to block the P-type channels, which have helped to decrease the occurrence of seizures. Overall, there are various non-selective calcium channel blockers that help alleviate symptoms of hypertension, schizophrenia, cardiac arrhythmia, epilepsy, pain, asthma, bradycardia, angina pectoris and Alzheimer's disease. Although many of the therapeutic compounds' main target is not P-type channels, further research needs to determine if the clinical effects of these compounds are also influenced by the P-type channel blockage.[1]
Related diseases
There are a number of neurological diseases that have been attributed to malfunctioning or mutated P/Q type channels.[6]
Alzheimer's disease
In Alzheimer Disease, there is a progressive accumulation of β-amyloid protein (Aβ) in brain. Amyloid plaques develop which result in the key symptoms of Alzheimer Disease. Aβ globulomer protein is an artificial substance used in research experiments that has similar properties to Aβ oligomer which is present in the body. Aβ oligomer directly regulates P/Q type calcium channels. The α1A subunit is the responsible for the conduction of calcium current. When only P/Q type calcium channels are present with Aβ globulomer protein, there is a direct effect on the α1A subunit and results in an increased calcium current through the P/Q type calcium channel. The response is dose dependent as concentrations of 20nM and 200nM of Aβ globulomer are necessary for significant increase of calcium current through channel in Xenopus oocytes, showing that a certain buildup of Aβ globulomer is necessary before the effects are seen. When the calcium current is increased, neurotransmitter release also rises, offering a possible cause for the toxicity in Alzheimer Disease patients.[9]
Migraine headaches
The CACNA1A gene codes for the alpha subunit of the P/Q type calcium channel.[10] The R192Q mutation of the CACNA1A gene is a gain of function mutation for P2X3 receptors.[5][10] P2X3 receptors are present in trigeminal ganglion neurons[5] and are believed to be a main contributor to familial hemiplegic migraine.[11] By using a knockin experiment, this mutation could be expressed in mice so research could be conducted.[5][10] The mutant mouse has a significantly higher P2X3 receptor activity than the wild type mouse[5] due to increased channel open probability and channel activation at lower voltages.[10] This increased receptor activity results in a higher flux of calcium through the P/Q type calcium channel. The increased intracellular calcium concentration may contribute to the acute trigeminal pain that typically results in a headache.[5] Evidence supports that migraines are a disorder of brain excitability characterized by deficient regulation of the cortical excitatory–inhibitory balance.[10]
Seizures
Levetiracetam is an anti-epileptic drug than can be used to treat partial and generalized seizures. Levetiracetam inhibits P/Q channel-mediated glutamate release and decreases the excitatory post synaptic currents of both AMPA and NMDA receptors in the hippocampus, specifically the dentate gyrus, which is known to propagate seizure activities. The inhibition of glutamate release results in an anti-epileptic response in patients because it decreases the excitatory postsynaptic current. There are many different types of calcium channels, so to prove that the P/Q type calcium channels are directly involved, a P/Q type voltage gated calcium channel inhibitor, omega-agatoxin TK, was used to block the channel. When blocked, patients no longer benefited from the anti-epileptic effects from the drugs. When blockers for L type and N type calcium channels were used, the effects of Levetiracetam were still seen. This is strong evidence that the P/Q type calcium channels are involved in the Levetiracetam treatment which allow for relief from seizures.[12]
Mutation studies
Many P-type calcium channels mutations result in a decreased level of intracellular free calcium. Maintaining calcium homeostasis is essential for normally functioning neurons. Changing the cellular calcium ion concentration acts as a trigger for multiple diseases, in severe cases these diseases can result in mass neuronal death.[6]
Mutation studies allow experimenters to study genetically inherited channelopathies. A channelopathy is any disease that results from an ion channel with malfunctioning subunits or regulatory proteins.[6] One example of a P-type calcium channel channelopathy is shown in homozygous ataxic mice, who are recessive for both the tottering and leaner genes. These mice present with mutations in the alpha1A subunit of their P/Q type channels. Mutations in these channels result in deficiencies within the cerebellar Purkinje cells that dramatically reduce the channels current density.[6]
The tottering mutations within mice result from a missense mutation and causes delayed-onset of seizures and ataxia. The tottering mutation substitutes a single proline instead of a leucine within the P-region of the channel. The P-region is responsible for the formation of the ion channel pore. The leaner mutation, which results in more severe symptoms than the tottering mutation, has been shown to result from a single nucleotide substitution that causes splicing failures within the channels open reading frame.[6] Mutations in the pore forming subunit of P type calcium channels cause ataxia, severely altered respiration, by decreasing minute ventilation and producing symptoms associated with atelectasis. Mutations to CaV2.1 have also been shown to affect transmission within the pre-Bötzinger Complex, a cluster of interneurons in the brainstem which help to regulate breathing.[5]
See also
References
- Nimmrich V, Gross G (October 2012). "P/Q-type calcium channel modulators". Br. J. Pharmacol. 167 (4): 741–59. doi:10.1111/j.1476-5381.2012.02069.x. PMC 3575775. PMID 22670568.
- Llinás R, Sugimori M (August 1980). "Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices". J. Physiol. 305: 171–95. doi:10.1113/jphysiol.1980.sp013357. PMC 1282966. PMID 7441552.
- Llinás R, Sugimori M, Lin JW, Cherksey B (March 1989). "Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison". Proc. Natl. Acad. Sci. U.S.A. 86 (5): 1689–93. doi:10.1073/pnas.86.5.1689. PMC 286766. PMID 2537980.
- Currie KP (2010). "G protein modulation of CaV2 voltage-gated calcium channels". Channels (Austin). 4 (6): 497–509. doi:10.4161/chan.4.6.12871. PMC 3052249. PMID 21150298.
- Nair A, Simonetti M, Birsa N, Ferrari MD, van den Maagdenberg AM, Giniatullin R, Nistri A, Fabbretti E (August 2010). "Familial hemiplegic migraine Ca(v)2.1 channel mutation R192Q enhances ATP-gated P2X3 receptor activity of mouse sensory ganglion neurons mediating trigeminal pain". Mol Pain. 6: 1744-8069–6-48. doi:10.1186/1744-8069-6-48. PMC 2940876. PMID 20735819.
- Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K (December 1998). "Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel". J. Biol. Chem. 273 (52): 34857–67. doi:10.1074/jbc.273.52.34857. PMID 9857013.
- Hillman D, Chen S, Aung TT, Cherksey B, Sugimori M, Llinás RR (August 1991). "Localization of P-type calcium channels in the central nervous system". Proc. Natl. Acad. Sci. U.S.A. 88 (16): 7076–80. doi:10.1073/pnas.88.16.7076. PMC 52236. PMID 1651493.
- Ovsepian SV, Friel DD (January 2008). "The leaner P/Q-type calcium channel mutation renders cerebellar Purkinje neurons hyper-excitable and eliminates Ca2+-Na+ spike bursts". Eur. J. Neurosci. 27 (1): 93–103. doi:10.1111/j.1460-9568.2007.05998.x. PMID 18093175. S2CID 43182491.
- Mezler M, Barghorn S, Schoemaker H, Gross G, Nimmrich V (March 2012). "A β-amyloid oligomer directly modulates P/Q-type calcium currents in Xenopus oocytes". Br. J. Pharmacol. 165 (5): 1572–83. doi:10.1111/j.1476-5381.2011.01646.x. PMC 3372738. PMID 21883149.
- Pietrobon D (July 2013). "Calcium channels and migraine". Biochim. Biophys. Acta. 1828 (7): 1655–65. doi:10.1016/j.bbamem.2012.11.012. PMID 23165010.
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR (1996). "Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4". Cell. 87 (3): 543–52. doi:10.1016/s0092-8674(00)81373-2. hdl:1765/57576. PMID 8898206. S2CID 16840573.
- Lee, Chun-Yao; Chin-Chuan, Horng-Huei (2009). "Levetiracetam Inhibits Glutamate Transmission through Presynaptic P/Q-type Calcium Channels on the Granule Cells of the Dentate Gyrus". British Journal of Pharmacology. 158 (7): 1753–1762. doi:10.1111/j.1476-5381.2009.00463.x. PMC 2801216. PMID 19888964.